您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2025-08-23 16:15
摘 要 / Abstract
目的:系統性掌握藥品實驗室數據完整性領域的國內外監管機構和行業的相關要求和主要做法。方法:全面梳理國內外9 個藥品監管機構、國際組織及行業協會等發布的數據完整性規范、指南、技術報告和問答,歸納各自的主要特點和應用方向,并對歷史版本的演變內容進行介紹。對數據完整性的重要概念和主要做法結合工作實際提出解讀意見,對數據完整性最新的監管理念進行解讀,對信息化條件下的藥品實驗室數據完整性方面的實際問題進行分析并給出建議。結果:本文首次完整歸納了國內外9個藥品監管機構、國際組織及行業協會等發布的歷版數據完整性規范、指南、技術報告和問答等文件的時間表,詳細分析了各自的重點內容,相關文件已基本涵蓋了藥品實驗室主要分析領域。同時,對目前仍容易產生分歧的數據完整性概念理解和工作做法進行了明確。此外,結合工作經驗提出了目前藥品實驗室審計追蹤的注意事項、實驗室數據采集滿足數據完整性屬性的注意事項、實驗室數據歸檔注意事項、實驗室中動態數據和混合系統數據的保存注意點、實驗室數據管理軟件主要驗證考慮事項等方面的工作建議。結論:本文通過對數據完整性相關文件發展演變進行系統性闡述,提出了解決藥品實驗室數據完整性關鍵問題的思路,以期為構建數據完整性合規的藥品實驗室提供有益參考。
Objective: This study aims to systematically examine the requirements and practices of domestic and international regulatory agencies and industry stakeholders in ensuring data integrity within pharmaceutical laboratories. Methods: A comprehensive review was conducted of data integrity standards, guidance, technical reports, and Q&A documents published by domestic and international drug regulatory authorities, international organizations, and industry associations. Key characteristics and areas of application were summarized, along with a historical overview of their evolution. Core concepts and best practices were interpreted in light of practical experience, and the latest regulatory perspectives on data integrity were analyzed. Common challenges faced under increasingly digital laboratory conditions were explored, with recommendations proposed. Results: This paper is the first to comprehensively compile a timeline of key data integrity documents published by nine major regulatory and professional bodies. It provides a detailed analysis of their main focus areas, covering essential aspects of laboratory analysis. At the same time, clarification is provided on frequently misunderstood concepts divergent practices. In addition, based on field experience, the paper presents practical recommendations for audit trail practices, data collection strategies that align with data integrity principles, data archiving, handling of dynamic and hybrid data systems, and software validation considerations. Conclusion: This paper provides a systematic explanation of the evolution of data integrity requirements, offering insights into addressing key issues related to data integrity in drug laboratories. It serves as a valuable reference for establishing compliantand robust data integrity management systems in pharmaceutical laboratories.
關 鍵 詞 / Key words
數據完整性;藥品監管機構;行業協會;規范;指南
data integrity; drug regulatory authority; industry association; regulatory standards; guidance
數據完整性(data integrity)是藥品全生命周期管理的重要組成部分,是藥品質量與安全的重要保障。在國內外藥品監管機構對藥品生產企業的合規檢查中,數據完整性問題是主要問題之一,其中實驗室環節發生的數據完整性問題在藥品生產企業數據完整性問題中占主要部分[1-4]。隨著藥品實驗室信息化進程加速,計算機化系統[5]、電子實驗筆記本[6]等工具廣泛應用,對數據完整性的管理要求已從傳統紙質記錄延伸至電子數據全生命周期。目前,全球多個藥品監管機構均將數據完整性作為重點監管領域,并相繼發布多份行業規范和實施指南等文件[7-8]。在確保數據完整性要求的前提下,這些文件的傾向和側重點均存在差異,甚至部分觀點也存在差異。例如,部分文件側重于數據完整性的總體要求和概念解釋,有些則側重于提供藥品實驗室具體檢查使用指導和微生物專門實驗室指導;部分文件僅涉及良好生產質量管理規范(good manufacturing practice,GMP)、良好供應和管理規范(good distribution practice,GDP)領域,有些則涵蓋GxP( 包括GMP、GDP、GCP 等)領域;部分文件強調對具體數據的細節要求,有些則強調從全生命周期以及數據完整性文化的角度進行考量。此外,部分文件在數據審計追蹤方面的要求也有所不同,并且隨著版本的更新,其監管思路亦呈現動態演進特征。現有研究已對數據完整性領域相關行業規范和指南等展開解讀[8-11],但仍存在完整的各版本歷史演變解讀不全、行業規范和指南等文件之間的綜合理解和解讀較少、藥品實驗室各分析領域側重點的解讀缺乏、數據完整性要求的最新發展解讀不足等問題。基于此,筆者從藥品實驗室角度出發,對比分析了國內外9 個藥品監管機構、國際組織及行業協會等發布的數據完整性規范和指南等文件的主要特點,并對目前藥品實驗室存在的數據完整性關鍵問題進行了初步探討,以期通過系統性闡述數據完整性發展演變歷程,精準把握相關文件的核心要義。同時,結合筆者工作經驗,針對藥品實驗室在數據完整性方面存在的主要問題提出解決思路,從而為構建符合數據完整性合規要求的藥品實驗室提供有益參考。
01數據完整性的歷史和定義
1.1 數據完整性監管的歷史沿革
數據完整性問題并非近年才凸顯的監管議題。早在1963 年,美國食品藥品監督管理局(Food and Drug Administration,FDA)就為了解決數據完整性問題,發布了《美國聯邦法規》(Code of Federal Regulations,CFR)第21 篇第133 條款(以下簡稱21CFR 133),該條款在1975 年變更為21 CFR 210《制造、加工、包裝或者保存藥品的優良制造規范》(Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding of Drugs; General) 和21 CFR 211《動態藥品生產管理規范》(Current Good Manufacturing Practice for Finished Pharmaceuticals) 兩部分。1997 年8 月20 日,FDA正式發布21 CFR 11《電子記錄與電子簽名》(Electronic Records; Electronic Signatures),這是首個針對電子數據管理的法規。值得注意的是,21 CFR 11 中未直接定義“數據完整性”這一術語,而是以“記錄完整性”的概念來替代。
1.2 數據完整性與數據可靠性
數據完整性和數據可靠性是data integrity 的不同中文譯法。2016 年10 月原國家食品藥品監督管理總局發布了《藥品數據管理規范(征求意見稿)》(2016 年版)[12],首次提出“數據可靠性(Data Integrity)” 并對其進行定義:數據可靠性是指貫穿整個數據生命周期的數據采集是完整的、一致的和準確的程度。所收集的數據應該是可歸屬的,清晰的,同步記錄的,原始的或真實副本,并且是準確的。2017 年8月發布的《藥品數據管理規范(征求意見稿)》(2017 年版)[13],對數據可靠性的定義進行了完善:數據可靠性指在數據生命周期內,數據完整、一致、準確的程度。應當以安全的方式收集和維護數據,從而保證數據歸屬至人、清晰可溯、同步記錄、原始一致、準確真實(國際上,常用縮略詞“ALCOA”或“ALCOA+”概括)。
數據完整性的定義最早見于英國藥品和健康產品管理局(Medicines and Healthcare Products Regulatory Agency,MHRA)2015 年3 月發布的《GMP 數據完整性定義和行業指南》(GMP Data Integrity Definitions and Guidance for Industry)[14], 其中指出數據完整性(data integrity) 是所有數據在整個數據生命周期中的全面、一致和準確的程度。MHRA在2 0 1 8 年發布的《GxP 數據完整性指南和定義》(‘GXP’ Data Integrity Guidance and Definitions)[15] 中指出, 數據完整性是數據的完整、一致、準確、可信和可靠程度,以及數據生命周期中數據這些屬性得到維護的程度。數據應以安全方式收集和保存,從而保障數據歸屬至人、清晰可溯、同步記錄、原始(或真實副本)和準確。世界衛生組織(World Health Organization,WHO) 在2016 年6 月發布的《良好數據和記錄管理實踐指南》(Guidance on Good Data and Record Management Practices)[16]中指出,數據完整性是指數據完整、一致、準確、可信和可靠的程度, 以及在整個數據生命周期中保持這些數據特征的程度。數據應以安全的方式收集和保存, 從而保障數據歸屬至人、清晰可溯、同步記錄、原始或真實副本及準確。綜合來看,MHRA 和WHO 均在相關文件中指出,數據完整性要求數據具有歸屬至人(attributable)、清晰可溯(legible)、同步記錄(contemporaneous)、原始一致(original) 和準確真實(accurate)的屬性(即ALCOA),以及完整(complete)、一致(consistent)、持久(enduring)和可及(available)的屬性(即ALCOA+)[15,17]。
考慮到國家藥品監督管理局(National Medical Products Administration,NMPA)于2020 年7 月發布的《藥品記錄與數據管理要求(試行)》[18] 中已經不再使用數據可靠性一詞,同時結合國內外相關行業對不同譯法的接受程度,本文暫采用數據完整性進行表述。
02國內外數據完整性要求特點及相關文件
筆者收集并梳理了1997 年以來國內外9 個藥品監管機構、國際組織及行業協會等發布的關于數據完整性的關鍵性規范、指南、技術文件等,詳細信息見表1。
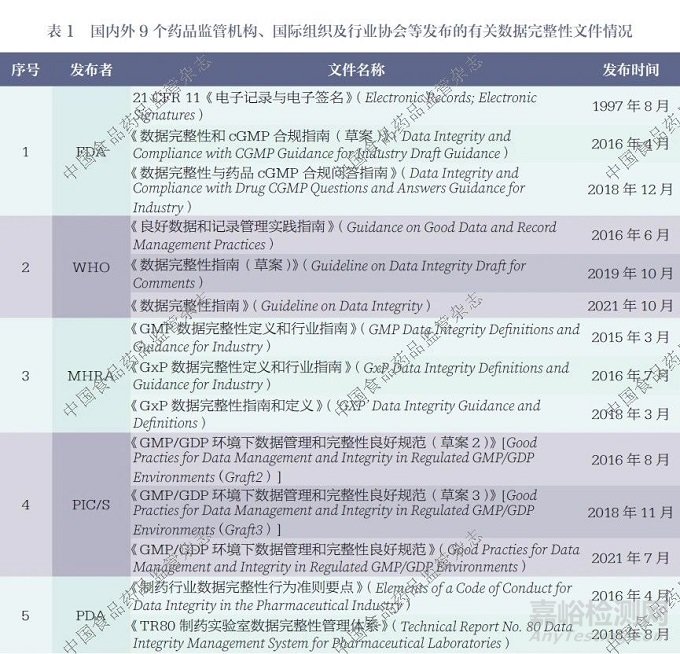

2.1 美國食品藥品監督管理局
目前FDA 尚未專門針對數據完整性問題發布完整且獨立的指南文件,但在其相關法規要求和行業指南等文件中,已有部分內容涉及數據完整性管理的原則性規定。2018 年12 月,FDA 發布了《數據完整性與藥品 cGMP合規問答指南》(Data Integrity and Compliance with Drug CGMP Questions and Answers Guidance for Industry)[19],該指南闡述了FDA 對于制藥企業在數據完整性方面重點關注且容易存在缺陷的18 個問題的態度,特別是對一些共性問題進行了詳細論述,包括關于計算機系統公用登錄賬戶問題,單機計算機設備保存紙質打印件或靜態記錄而非原始記錄的問題, 在“系統適用性”測試或試檢、預檢、平衡運行中使用實際樣品的問題,以及僅保留重新處理的實驗室色譜最終結果而未保留原始數據的問題等。FDA 認為,數據完整性問題屬于21 CFR210、21 CFR 211 和21 CFR212 所規定的《現行良好生產規范》(Current Good Manufacturing Practice,cGMP) 基本問題。ALCOA 原則的所有重要屬性在cGMP 中均有相應的監管依據,并且FDA 針對每個問題都給出了cGMP 條款參考。基于此,FDA認為21 CFR 11、21 CFR 210、21 CFR 211 和21 CFR 212 已對制藥企業的數據完整性提出了實質性要求,因此沒有必要再發布專門的指南文件。
2.2 英國藥品和健康產品管理局
2015 年3 月,MHRA 發布《GMP 數據完整性定義和行業指南》(GMP Data Integrity Definitions and Guidance for Industry)[14], 該指南介紹了MHRA 對于數據完整性的立場,明確了達成合規要求的最低期望,并提出了基于風險的數據管理方法,包括數據風險、數據關鍵程度和數據生命周期等方面。這是首個涵蓋了數據完整性相關術語定義和要求的指南,為正確理解數據完整性問題提供了具有積極意義的參考。此后,MHRA分別在2016 年7 月和2018年3 月發布了《GxP 數據完整性定義和行業指南》(GxP Data Integrity Definitions and Guidance for Industry)[20] 和《GxP 數據完整性指南和定義》[15]。在這2 份指南文件中, 數據完整性的適用范圍從制藥行業擴展至GxP 領域,但不包括醫療器械和藥物警戒領域。同時, 指南文件明確指出, 其主要涉及數據完整性,而不是數據質量,ALOCA+ 中的“+” 是為了強調相關要求而添加的,所以無論是ALCOA 還是ALCOA+, 其在數據完整性的期望方面并無差異。由于英國是經濟合作與發展組織(Organization for Economic Co-operation and Development,OECD) 的成員國,因此在良好實驗室管理規范(good laboratory practice,GLP)領域,OECD 發布的《良好實驗室規范工作組關于GLP數據完整性咨詢文件》(Advisory Document of the Working Party on Good Laboratory Practice on GLP Data Integrity)[21] 具有更高的優先級,其效力優于MHRA 的相關指南。
2.3 歐洲藥品管理局
與FDA 類似, 歐洲藥品管理局(European Medicines Agency,EMA)也未發布專門的數據完整性指南,而是采用問答的形式,闡述了其對數據完整性的監管理念。2016 年8 月,EMA 在“良好生產規范和良好銷售規范指南: 問題和回答”欄目中,增加了對數據完整性方面23 個問題的回復內容。EMA 指出,要通過國際人用藥品注冊技術協調會(ICH)發布的《Q9 :質量風險管理》(Q9: Quality Risk Management) 中的方法, 對數據全生命周期的各個階段開展數據風險性和數據關鍵性評估,并在此基礎上,采用相應的控制措施來實現最終的數據完整性目標。EMA 同時強調了數據完整性的監管期望是數據需要符合ALCOA原則,并提供了每項ALCOA 原則與歐盟GMP 參考條款的對應關系,表明歐盟GMP 條款能夠滿足數據完整性的管理要求。
2.4 中國國家藥品監督管理局
2016 年10 月和2017 年8 月先后發布的《藥品數據管理規范(征求意見稿)》的2 個版本,提出了數據可靠性的概念并進行了修訂。在這2 個版本中,規范文件的適用范圍從“藥品研制、生產、流通等活動,包括從事上述活動的臨床試驗、合同研究(CRO)、委托生產(CMO)、委托檢驗等單位和個人”,修訂為“藥品研發、生產、流通、上市后監測與評價等產品生命周期中全部活動的數據管理”。同時,數據的管理原則從“數據管理應貫穿整個數據生命周期,堅持真實、準確、及時、可追溯的數據管理原則,確保數據可靠性(Data Integrity)”,修訂為“數據管理應當遵守歸屬至人、清晰可溯、同步記錄、原始一致、準確真實的基本要求,確保數據可靠性”,并對數據各項ALCOA 屬性進行了相應的管理規定,強調了數據的全生命周期管理理念。不過,該規范的正式版本至今尚未發布。
NMPA 在2020 年7 月發布了《藥品記錄與數據管理要求(試行)》[18], 該要求自2020 年12月1 日起施行。其中明確指出,其適用范圍為在我國境內從事藥品研制、生產、經營、使用活動中產生的,應當向藥品監督管理部門提供的記錄與數據。同時,該要求對記錄和數據的概念進行了清晰界定,并分別針對紙質記錄和電子記錄制定了相應的規定,這些規定主要傾向于原則性的管理要求。
2.5 世界衛生組織
WHO 在2016 年6 月發布了《良好數據和記錄管理實踐指南》,此后,該指南歷經更新,于2019 年10 月和2021 年10 月分別被《數據完整性指南(草案)》(Guideline on Data Integrity Draft for Comments)[17] 和《數據完整性指南》(Guideline on Data Integrity)[22] 所取代。WHO 的指南較早地對數據完整性進行了定義,但隨著版本不斷更新和升級,至《數據完整性指南》發布時,已不再對數據完整性進行定義解釋,而只保留了對數據完整性ALCOA+ 屬性要求的相關內容, 這體現了其指導思想的轉變。除了強調數據全生命周期管理和質量風險管理的重要性之外,WHO 還特別強調了管理層在數據完整性方面應承擔的職責和企業質量文化所產生的影響,同時提出了對紙質數據的優良文件規范要求以及對計算機化系統完整性的要求等。尤其值得關注的是,指南附錄1 中列舉了11 個數據完整性管理案例,這些案例對正文中的數據完整性要求起到了輔助說明的作用。同時,針對紙質數據、電子數據和混合系統數據的各項ALCOA 屬性要求,案例也均進行了具體細化,具有較強的指導意義。
2.6 藥品檢查合作計劃
藥品檢查合作計劃(Pharmaceutical Inspection Co-operation Scheme,PIC/S)是一個由各國(地區)藥品監管機構在GMP 領域組成的非正式組織。該組織的主要職責是制定GMP 領域的通用標準,并為檢查員提供相關培訓機會,因此其工作重點側重于GMP 檢查員角度。PIC/S 分別于2016 年8 月、2018 年11 月和2021 年7 月發布了《GMP/GDP 環境下數據管理和完整性良好規范》(Good Practies for Data Management and Integrity in Regulated GMP/GDP Environments)的第2 稿草案[23]、第3 稿草案[24] 以及定稿指南[25],版本的升級主要是在計算機化系統方面完善了數據傳輸和混合系統的完整性相關要求。PIC/S 強調數據要符合ALCOA+原則,并且列舉了ALCOA+ 各原則與該指南條款的對應關系。從數據全生命周期的范圍出發,PIC/S 基于質量風險管理角度,對紙質系統、計算機化系統和混合系統的數據完整性提出了具體要求,并且從檢查的視角提出了各環節未達到預期時應關注的風險點。此外,PIC/S 還強調了質量文化在保障數據完整性方面發揮的重要作用,并且提出了對外包活動的數據完整性要求。同時,PIC/S 對數據完整性缺陷進行了分類,并給出了解決數據完整性問題的具體方法流程。
2.7 美國注射劑協會
美國注射劑協會(Parenteral Drug Association,PDA) 在2016 年4 月發布的《制藥行業數據完整性行為準則要點》(Elements of a Code of Conduct for Data Integrity in the Pharmaceutical Industry)[26],對制藥行業數據完整性的關鍵點進行了概述,例如,數據收集、分析、報告和保存的要求,電子數據采集系統要求,員工培訓要求,數據完整性問題的上報流程以及對問題行為的調查等。PDA 在2018 年8 月發布了《TR80 制藥實驗室數據完整性管理體系》(Technical Report No. 80 Data Integrity Management System for Pharmaceutical Laboratories)[27]技術報告,該報告提出了藥品監管機構對數據完整性的要求和期望,并將人為因素作為質量控制(quality control,QC) 實驗室數據完整性問題的考慮要素之一。同時,該報告還系統論述了微生物實驗室和QC 實驗室數據完整性的管理要求,特別是考慮到微生物檢測以手工操作為主的數據完整性特點,分析了實驗室混合系統、計算機化系統和紫外- 可見分光光度儀、紅外分光光度儀等其他儀器設備的數據完整性要求,以及實驗室數據管理軟件在數據完整性方面的要求。此外,PDA 還建議將ICH Q9 的質量風險管理理念應用于數據治理中,并提出了對數據完整性方面的漏洞進行糾正的方法。該報告為藥品實驗室在數據完整性管理方面提供了具體且實用的操作方法。
2.8 國際制藥工程協會
國際制藥工程協會( International Society for Pharmaceutical Engineering,ISPE) 在2016 年6 月發布了《電子記錄與數據完整性指南(草案)》(Electronic Records and Data Integrity Graft), 并于2017年3 月正式發布了《記錄與數據完整性指南》(Records and Data Integrity Guide)[28]。ISPE 強調,數據應符合ALCOA+ 原則,對于數據完整性的管理應是基于ICH Q9 質量風險管理理念的數據全生命周期治理。該指南指出了數據治理需要充分考慮人為因素,并且在涵蓋管理、發展、操作等內容的14 個附錄中提供了具體的數據完整性操作指導。值得注意的是,ISPE 還引入了數據完整性成熟度評級方法。該方法基于文化、治理與組織、戰略規劃和數據完整性計劃、法規、數據生命周期、數據生命周期支持過程等6 個方面共36 個評價因素,對企業的數據可靠性進行5 個等級評價,等級從低到高依次為一級至五級。這一評估方式能夠幫助企業找到自身在數據完整性管理方面存在的不足,并為其指明提升數據完整性水平的最佳方向。
2.9 歐洲原料藥委員會
歐洲原料藥委員會(Active Pharmaceutical Ingredients Committee,APIC) 分別于2019 年3 月和2022 年4 月發布了《基于風險的數據完整性管理實踐指南》(Practical Risk-Based Guide for Managing Data Integrity)的第1 版[29] 和第2 版[30]。相較于第1 版,第2 版指南主要新增了審計追蹤管理相關內容,補充了中級和中高級嚴重性數據來源和例子,并將示例重新命名為附件,移至獨立的附件文檔中。該指南著重介紹了一種全面的數據完整性風險管理方法,具體而言,它依據數據全生命周期的特點對數據進行分類管理,明確各類數據對應的系統最低要求;同時,系統性地評估數據管理中各體系和流程存在的差距,進而開展風險評估,并根據風險的嚴重程度制定相應的風險管理措施。此外,該指南采用業務流程圖和工作表的形式,提供了一系列實用的數據完整性風險管理工具。2023 年4 月,APIC 發布了一份數據完整性常見問題的技術資料,針對數字和電子簽名、密碼管理、訪問管理以及記錄的生命周期管理等4 個方面的11 個相關問題給出了指導意見。
對于上述發布的數據完整性規范、指南、技術報告等文件,筆者對部分文件的涵蓋范圍、數據完整性屬性、數據完整性涉及要素內容以及重點關注內容進行了統計分析,詳見表2。

03數據完整性相關重要概念及關系
3.1 數據與記錄的關系
數據與記錄有著密不可分的關系。NMPA 發布的《藥品記錄與數據管理要求(試行)》中指出,“數據是指在藥品研制、生產、經營、使用活動中產生的反映活動執行情況的信息,包括:文字、數值、符號、影像、音頻、圖片、圖譜、條碼等;記錄是指在上述活動中通過一個或多個數據記載形成的,反映相關活動執行過程與結果的憑證。”由此可見,數據是藥品相關活動執行過程中產生的信息,而記錄是反映藥品相關活動執行情況的憑證。簡言之,記錄是數據的憑證化呈現,數據是記錄所承載的信息內容,兩者在實質內容上是一致的,只是在載體形式上有所區別。
3.2 完整性數據的屬性特征
國內外藥品監管機構、國際組織及行業協會等對數據完整性的認識基本一致, 普遍認為數據應該具備ALCOA 屬性, 甚至需達到ALCOA+ 屬性標準。ALCOA 屬性的具體特征為:可追溯的(attributable), 即能夠追溯到數據生成的具體個人,并在必要時追溯到測量系統;可讀的(legible),即數據和元數據在數據的整個生命周期內應可讀;同步的(contemporaneous),即相關人員應在數據和信息生成或獲取時立即進行記錄;原始的(original),即原始記錄(或經驗證的真實副本),是首次獲取的數據或信息,以及為全面重建GxP 活動實施過程所需的所有后續數據,均應可獲取;準確的(accurate), 即數據是正確的、真實的、完整的、有效的和可靠的。ALCOA+ 屬性是在ALCOA 屬性的基礎上,進一步增加了CCEA 特征,具體含義為:完整的(complete), 即數據必須是一個完整的集合;一致的(consistent),即數據必須相互一致;持久的(enduring),即數據是耐用的,并且貫穿整個數據生命周期;可獲取的(available),即數據隨時可用于審查或檢查目的。ALCOA+ 屬性主要是從數據使用需求的角度,進一步強化了對數據完整性的要求,但其本質上與ALCOA 屬性沒有差別,都是為了確保數據的質量和可靠性。
3.3 數據保存、備份、歸檔、傳輸和遷移
在數據完整性的相關規范和指南等文件中,對于數據保存、備份、歸檔、傳輸和遷移這幾個名詞的解釋基本類似。數據保存是以歸檔(針對需要長期存儲而受保護的數據)或備份(為應對災難恢復而保存的數據)為目的的數據處理過程。數據備份是在規定的時間間隔內,以安全的方式復制實時電子數據,以確保在需要時數據能夠被成功恢復。數據歸檔是在規定的保存期限內,對記錄進行長期存儲和保護的過程,目的是防止其變質、被篡改或刪除。此外, 歸檔還包括將電子記錄從活動數據庫中刪除。數據傳輸是在不同的數據存儲類型、格式或計算機化系統之間傳輸數據的過程。數據遷移是將已存儲的數據從一個持久的存儲位置轉移至另一個存儲位置的過程。
數據的保存、備份、歸檔、傳輸和遷移都需要經過嚴格驗證,以確保其滿足數據完整性的要求。具體而言,數據備份是短期行為,旨在應對突發情況下的數據恢復;而數據歸檔是長期行為,側重于數據的長期保存和保護。PDA、ISPE 和PIC/S 指出,備份數據不應就地存檔,而是應存于距離較遠(物理分離)的位置。對于數據傳輸,PIC/S 發布的《GMP/GDP 環境下數據管理和完整性良好規范》中特別指出,數據傳輸要確保數據直接傳輸到安全的位置和(或)數據庫,而不是簡單地從本地驅動器復制,這是因為本地復制存在數據被篡改的風險。數據遷移過程中可更改數據的格式,但不應更改內容或含義,且遷移后原存儲地址不再保留原始數據。
3.4 混合系統數據管理要點
混合系統是指一種集數據管理和控制功能于一體的系統,通常由生成電子數據的電子系統和生成紙質記錄的人工系統組成。因此,來自混合系統的完整數據組會同時包括電子數據和紙質數據。ISPE 發布的《記錄與數據完整性指南》以及PIC/S 發布的《GMP/GDP 環境下數據管理和完整性良好規范》均明確指出紙質流程和電子流程之間的接口是混合系統數據完整性的風險點,強調應建立相應的程序并做好記錄,以控制手動系統和自動系統之間的接口,尤其是人工生成的數據手動錄入計算機系統、將自動化系統生成的數據轉錄(包括手動轉錄)到紙質記錄上,以及自動檢測打印數據并將其轉錄到計算機化系統中等。此外,還需要注意的是,紙質原始記錄經過掃描生成電子副本后,盡管該副本可被認定為真實副本,但對于紙質記錄的處置仍需謹慎,必須遵循相關法律法規、監管要求以及機構內部管理制度等的要求,來決定紙質記錄是否可以被銷毀,并嚴格執行必要的管理程序。
3.5 對數據完整性的理解
結合國內外藥品監管機構、國際組織及行業協會等對于數據完整性要求的理解,實現數據完整性就是要在數據的全生命周期內,建立一套基于風險評估的完善的數據治理體系,包括良好的文件管理規范以及數據完整性文化氛圍,以此確保數據始終具備ALCOA+ 屬性特征。數據完整性工作不存在唯一的標準做法,而是需要與各自的實際工作相結合。只有深入理解并合理運用上述概念, 尤其是準確把握ALCOA+的內涵,才能真正達到數據完整性的目標。
3.6 審計追蹤的范圍
審計追蹤作為元數據的一種表現形式,包含了與GxP 記錄的創建、修改或刪除等操作相關的信息。審計追蹤是實現數據完整性的基本要求,無論是紙質系統還是電子系統,均需滿足這一要求;在具備條件的計算機化系統中,其更是必要條件。對于計算機化系統中審計追蹤范圍的理解,MHRA 在其發布的《GxP 數據完整性指南和定義》中指出,審計追蹤不必要包括每個系統活動,如用戶登錄或退出、敲鍵等操作無需記錄在審計追蹤中;FDA 發布的21 CFR 11 第11.10 (e) 節要求,使用安全的、計算機生成的、帶有時間戳的審計追蹤,獨立記錄操作員輸入的日期和時間,以及創建、修改或刪除電子記錄的操作;ISPE 發布的《記錄與數據完整性指南》進一步闡釋,FDA這一要求特別針對操作員的輸入和操作,包括創建、修改或刪除受監管的電子記錄,但不包括用戶執行的所有活動,也不包括所有系統操作;APIC 發布的《基于風險的數據完整性管理實踐指南》中提出,應對關鍵數據進行審計追蹤記錄,其中關鍵數據指的是那些可能影響產品質量的數據。從上述描述來看,審計追蹤主要是針對影響關鍵數據的信息進行追蹤,并非對全部行為活動的信息都進行追蹤。
3.7 質量文化因素對數據完整性的重要性
當前,實驗室質量文化因素對數據完整性的重要性正日益受到關注。WHO 在《數據完整性指南》中指出,高級管理層有責任營造一個有利于建立、保持和持續改進質量文化的環境,并且支持透明和公開地報告組織各級偏差、錯誤或遺漏和數據完整性失誤等情況。PIC/S 發布的《GMP/GDP 環境下數據管理和完整性良好規范》中提出,質量文化是管理層、團隊負責人、質量人員以及所有有助于創建質量文化以確保數據質量和完整性的人員,所共同展示出的價值觀、信念、思維方式和行為模式的集合。ISPE 在《記錄與數據完整性指南》中強調,人為因素在數據完整性中有著關鍵性影響。人為因素涵蓋多個方面,包括公司和當地的文化、事故類型及原因、人為失誤、數據偽造和欺詐、不公正壓力以及適當的行為控制等。其中,公司和當地的文化影響是需重點考慮的因素之一。綜合上述描述可以看出,在實驗室中,構建自上而下全員參與的數據完整性質量文化體系,強化全員質量意識,并實施行之有效的數據完整性質量行為和質量機制,是確保數據完整性的關鍵要素。
04藥品實驗室數據完整性管理相關思考
針對藥品實驗室如何有效落實數據完整性相關要求這一問題,筆者根據國內外數據完整性相關規范和指南等文件,以藥品監管機構、國際組織及行業協會等提出的相關要求為基礎,對其中需要重點關注的事項進行了歸納,并提出如下思考。
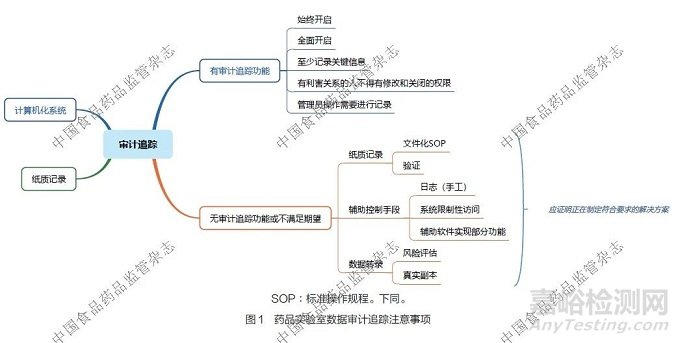
4.1 藥品實驗室數據審計追蹤的注意事項
對于實驗室中具備審計追蹤功能的計算機化系統,應全面且始終開啟審計追蹤功能,同時至少記錄關鍵操作信息。與數據有利害關系的人員不得有修改和關閉審計追蹤功能的權限,即使是信息技術管理員進行操作,也應進行詳細記錄。對于實驗室中不具備審計追蹤功能的計算機化系統,需采取有效的輔助控制手段,如建立日志(含手工記錄日志)、實施系統限制性訪問措施、借助輔助軟件實現部分審計追蹤功能等。對于實驗室的紙質記錄,應制定文件化的操作規范,并進行相關的操作驗證,以確保記錄過程的規范性和準確性。對于無審計追蹤功能的計算機化系統所涉及的數據轉錄操作,應該先進行數據完整性風險評估,并且要確保轉錄的數據為原始數據的真實副本。實驗室應將所有無審計追蹤功能或不滿足相關期望的系統納入改進計劃,制定符合要求的解決方案并推進實施。藥品實驗室數據審計追蹤的注意事項詳見圖1。
4.2 藥品實驗室數據采集滿足ALCOA+ 屬性的注意事項
藥品實驗室數據采集滿足ALCOA+ 屬性要求是保障數據完整性的根本。針對不同類型的實驗數據,筆者分別闡述了滿足相關要求的注意事項,詳見圖2。
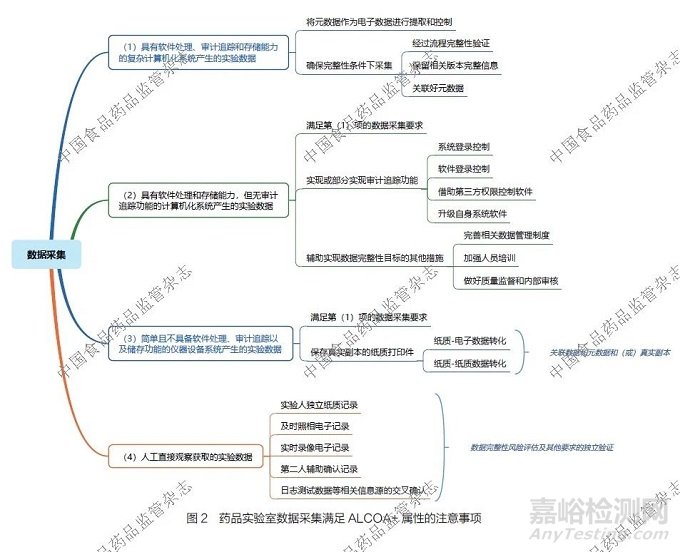
(1)具有軟件處理、審計追蹤和存儲能力的復雜計算機化系統產生的實驗數據。對于此類實驗數據,如液相色譜儀、氣相色譜儀以及各類質譜儀等產生的數據:①應盡最大努力將元數據作為電子數據進行提取和控制。②若數據以圖片、PDF 等固定電子格式形式采集,在經過流程完整性驗證、保留相關版本完整信息,以及關聯好元數據的前提下,可認可其具備數據完整性。
(2)具有軟件處理和存儲能力,但無審計追蹤功能的計算機化系統產生的實驗數據。對于此類實驗數據,如紫外- 可見分光光度儀、紅外分光光度儀、電位滴定儀等產生的實驗數據:①要盡量滿足第(1)項下關于數據的采集要求。②要通過系統登錄控制、軟件登錄控制,借助第三方權限控制軟件,以及升級自身系統軟件等措施,實現或部分實現審計追蹤功能,至少要實現關鍵權限控制功能。③完善相關數據管理制度,加強人員培訓,做好質量監督和內部審核等工作,也可以輔助實現藥品實驗室數據完整性的目標。
(3)簡單且不具備軟件處理、審計追蹤以及儲存功能的儀器設備系統產生的實驗數據。對于此類實驗數據, 如pH 計、天平、不溶性微粒分析儀等產生的實驗數據:①要盡量滿足第(1)項下關于數據的采集要求。②若儀器設備具備同步數據打印功能,且經過驗證打印數據是數據和元數據的真實副本時,可保存打印數據。由于部分儀器采用的是熱敏紙,需要采用經過驗證的程序進行紙質- 電子數據轉化或者紙質-紙質數據轉化,以實現長期保存,并盡量與元數據和(或)真實副本有效關聯。
(4)人工直接觀察獲取的實驗數據。對于此類實驗數據,如顯色、沉淀、產氣、顯微、比色和薄層等產生的實驗數據,在經過數據完整性風險評估及其他要求的獨立驗證后,可根據風險和要求的不同,采用以下1 種或多種數據采集形式:實驗人獨立紙質記錄、及時照相電子記錄、實時錄像電子記錄、第二人輔助確認記錄,以及日志測試數據等相關信息源的交叉確認等。
4.3 藥品實驗室數據歸檔注意事項
藥品實驗室在進行數據歸檔時,須遵循以下3 個原則:①對等原則。無論是電子數據還是非電子數據,均應同等重視并予以規范處理。②對象原則。歸檔的數據必須是原始記錄和(或)經過驗證的真實副本。③可用原則。歸檔的數據要在數據有效期內具備可檢索和可讀取等特性。藥品實驗室數據歸檔注意事項詳見圖3。

若實驗室數據系統進行升級,可能會出現遺留系統無法再支持數據讀取的情況。此時,可以根據對數據重要性的評估結果,制定分級的數據歸檔策略:①應優先考慮盡量維護原軟件系統。②若不能在原軟件系統中繼續維護,可以嘗試將原軟件系統遷移至虛擬環境中繼續進行維護。③若在虛擬環境中維護也不可行,則需將數據遷移至一個可繼續訪問的系統。該系統應處于受控狀態,且經過測試和驗證后,證明其能實現相應功能。④若實在無法找到合適的系統來維護數據,可將原始數據轉換為經過驗證的真實副本的替代文件(包含動態數據和靜態數據)進行歸檔。⑤作為最后手段,可對數據中的重要屬性部分進行驗證副本的轉化,然后進行歸檔。
4.4 藥品實驗室中動態數據和混合系統數據的保存注意點
實驗室中動態數據和混合系統數據的保存應遵守ALCOA+原則、根據預定用途原則和保存原始數據和元數據原則,其注意事項詳見圖4。

對于動態系統中的數據:①應在數據獲取的原始狀態下進行保留。②對于紙質打印保留的數據,要注意需包含所有元數據,且要保留動態數據的鏈接,或者經過驗證后保留重要屬性的副本,但紙質打印數據通常缺乏可編輯性,因此存在較高風險。③計算機化系統若無法繼續維持運行,則應保存經過驗證的副本。該副本應包含原始數據、元數據、審計追蹤和結果文件等,盡可能全面涵蓋重建所需的所有數據。④紙質打印數據和計算機化系統驗證副本的過程都要經過驗證,并形成文件化的操作規程。
對于混合系統的數據保存,需要對整個數據集(動態數據和靜態數據)進行完整保存,并且保存的過程同樣要經過驗證,形成文件化的操作規程。
4.5 藥品實驗室數據管理軟件主要驗證考量要點
在實驗室中,會使用到實驗室信息管理系統、電子實驗筆記本等數據管理軟件。為確保數據完整性,需要對這些系統進行適用性驗證,應重點關注以下要點:①記錄軟件名稱和版本號。②驗證軟件以確保其符合實驗室預期用途,包括整體符合需求性和質量體系具體符合需求性。③確認軟件的審計追蹤功能滿足相關法規要求。④確認軟件的網絡傳輸功能滿足實驗室實際傳輸需求,包括設備至交換器、設備至終端服務器、服務器和交換器至應用或數據服務器、應用服務器與數據服務器之間等多個環節。⑤服務器的有效性和安全性驗證。評估服務器的運算保障能力,包括應用服務器的運算處理能力、應用服務器的負載均衡、儲備和應急能力,以及應用服務器與數據服務器的匹配性等, 確保其能滿足實驗室數據管理的計算需求;評估并確認服務器的安全保障,包括配備相應防火墻,考慮殺毒軟件的兼容性,以及限制終端計算機系統使用移動存儲器等。⑥基于云系統的驗證考慮,包括驗證數據管理系統的安全性和泄露風險,建立基于云系統的質量服務協議和應急計劃,并確定服務器的確切物理位置,軟件和(或)系統應能恢復原始驗證狀態等。⑦對系統軟件使用的統計計算功能進行符合規范或標準要求的驗證程序,不同的實驗可以采用不同的驗證參數,如平均值、標準偏差、相對標準偏差和溶出度相似因子(f2)等。在驗證過程中,尤其要關注修約的次數和規則,及其對結果符合性判斷的影響,特別是對臨界數據的符合性判斷。實驗室數據管理軟件驗證注意事項詳見圖5。

05結 語
國內外藥品監管機構、國際組織及行業協會對于藥品實驗室數據完整性的要求,既存在共性,又各具特色,并且在不同的GxP領域內還有相對細化的具體要求。隨著對數據完整性認識的不斷深入,筆者也對處于不同發展階段的藥品實驗室在數據完整性方面提出了新的思考和完善相關體系的建議。具體到各實驗室,需要根據自身的實際情況進行綜合分析,切實理解并落實數據完整性的關鍵性原則做法,同時以文件化的形式,對重點環節的數據操作進行規范。其中,關鍵性原則做法就是要基于良好的數據完整性質量文化,在數據的全生命周期內,依托風險評估治理體系,確保數據具備ALCOA+ 全部屬性。

來源:中國食品藥品監管雜志


