糖尿病是一種以長期高血糖為特征的代謝紊亂疾病,是全球最常見的慢性病之一。國際糖尿病聯盟2019年發布的全球糖尿病數據顯示[1],全球共計有約7億成人糖尿病患者(20-79歲),每11個成人就有1人患有糖尿病(4.63億),成人糖尿病患者最多的國家依次是中國、印度和美國,其中中國成人糖尿病人數高達1.16億。
2019年,首個口服GLP-1受體激動劑(GLP-1 receptor agonist, GLP-1RA)——諾和諾德的口服索馬魯肽(Semaglutide)被FDA批準上市,口服的給藥方式可減少糖尿病人給藥的創傷,大大提高用藥依從性和便利性,有望改變糖尿病藥物市場格局。本文概述了重要靶點GLP-1受體的發現,索馬魯肽皮下注射制劑和口服制劑的開發和相應的藥代動力學特征。
一、GLP-1受體及GLP-1受體激動劑
胰高血糖素樣肽-1(Glucagon-like Peptide 1,GLP-1),是由三十個氨基酸組成,由人胰高血糖素基因編碼,腸道L細胞分泌的一種肽類激素。當GLP-1與胰島β細胞表面的GLP-1受體結合后,β細胞質內的鈣離子濃度升高,增強胞內胰島素貯藏顆粒的胞吐作用,促進胰島素釋放。GLP-1還可以刺激胰島β細胞的增殖和分化,抑制β細胞凋亡。然而,不是生物體內所有形式的GLP-1都具有降糖作用,體內有生物活性的GLP-1有兩種形式,即圖2中的GLP1 7-37和7-36。GLP-1會迅速被細胞表面的DPP-4蛋白酶水解,從而失去降糖活性,因而不具備穩定性。因此,針對GLP-1受體的降糖藥物不僅需要與GLP-1具有相同或者類似的生物活性,且應不易被DPP-4蛋白酶水解,即GLP-1受體激動劑(GLP-1 receptor agonist, GLP-1RA)。
 圖1: GLP-1的不同結構形式[2]
圖1: GLP-1的不同結構形式[2]
二、索馬魯膚皮下注射制劑的開發
索馬魯肽(Semaglutide)由諾和諾德研發,最初被設計為一種強有力的、持久的皮下注射類抗糖尿病藥物,其用藥頻率為每周一次,以提高便利性。索馬魯肽于2017年12月獲FDA批準上市,是繼艾塞那肽、利拉魯肽、阿必魯肽、度拉糖肽、利司那肽、貝那魯肽之后,全球第七個獲批的GLP-1RA。
作為諾和諾德的第二款GLP-1RA藥物,索馬魯肽的研發策略借鑒了公司第一款GLP-1RA藥物利拉魯肽,在內源GLP-1的序列結構上進行改變,并通過在肽鏈骨架上修飾脂肪酸側鏈與人白蛋白結合來延長血漿半衰期[3]。
具體來講,首先索馬魯肽以內源GLP-1的序列和結構為出發點,以降低免疫原性風險。隨后,通過丙氨酸掃描突變技術,研究人員發現,將內源GLP-1第8位丙氨酸替換為α-氨基異丁酸(一種非天然氨基酸),不僅能夠保持藥物與GLP-1受體結合親和力,同時還可以保護多肽在N端不被DPP-4降解。然而,進行脂肪酸修飾的實驗中,研究人員發現,GLP-1RA與白蛋白之間的強親和力會影響GLP-1RA與GLP受體的結合,因此如何平衡半衰期與藥效之間的關系是非常關鍵的問題。經過對連接子和脂肪酸進行大量的篩選和對比,最后確定在26號賴氨酸使用γGlu-2xOEG連接子和C18二元脂肪酸側鏈進行修飾,能夠同時保證白蛋白的高親和性和對GLP-1受體的高效力。另外,研究人員還用精氨酸替換第34位的賴氨酸,以防止脂肪酸側鏈連接到錯誤的位置。索馬魯肽的分子示意圖見圖3 [4],其對GLP-1受體的親和力為0.38± 0.06 nM,比利拉魯肽減弱三倍,但對白蛋白的親和性是利拉魯肽的6倍,在人體的半衰期長達165小時[5]。
圖2:索馬魯肽藥物分子結構示意圖(皮下給藥制劑)[4]
2019年9月,全球首個非注射型GLP-1RA藥物口服索馬魯肽片獲FDA批準上市,每日早餐前半小時空腹服用一次,用于結合飲食控制和運動,以改善Ⅱ型糖尿病成人患者的血糖控制。索馬魯肽口服制劑的出現改變了GLP-1RA藥物的給藥方式,它減少糖尿病人給藥的創傷,提高了用藥依從性和便利性。
索馬魯肽從皮下注射制劑到口服制劑,遠比想象中困難。胃腸道中豐富的蛋白酶,會迅速水解多肽,且多肽藥物分子量大,滲透性低,吸收困難,因此口服給藥難度極大,此前已上市的GLP-1RA藥物用藥方式多數為皮下注射。但是從方便病人服藥的角度來說,口服給藥是一種相當理想的遞送方法。通常口服藥物為小分子,需要具有一定的疏水性。對于GLP-1RA來說,一種方法是開發激活GLP-1受體的小分子激動劑,但小分子激動劑的效力不足、與GLP-1R結合缺乏特異性以及半衰期不理想。另一種策略是使用吸收增強劑。由Emisphere公司開發的N-[8-(2-羥基苯甲酰基)氨基]辛酸鹽(SNAC)是一種公認安全的小分子吸收促進劑(圖4),可以協助口服遞送藥物。SNAC在藥學領域并不活躍,主要用做食品補充劑、維生素和膳食成分。SNAC已經之前也用于和肝素、伊班膦酸鹽和維生素B12共同配制以增加藥物的吸收[6-8]。
圖3:N-[8-(2-羥基苯甲酰基)氨基]辛酸鹽的結構式
索馬魯肽作的口服制劑就是和SNAC一起構成的配方。肽類藥物主要在腸道中被吸收而口服索馬魯肽改變了肽的吸收部位,使其在胃部被吸收,而不是在腸道中。在吸收的過程中,SNAC在胃部的溶解能夠局部升高胃的pH值,進而提高索馬魯肽的溶解度,且將胃中酸性環境改變為中性環境,使肽酶失活,避免索馬魯肽被胃中的肽酶降解。實驗表明,SNAC的親脂性使其能夠嵌入到細胞膜上,幫助索馬魯肽被細胞快速吸收。索馬魯肽在胃中的溶解如圖5所示[9]。
圖4:索馬魯肽在胃中溶解示意圖
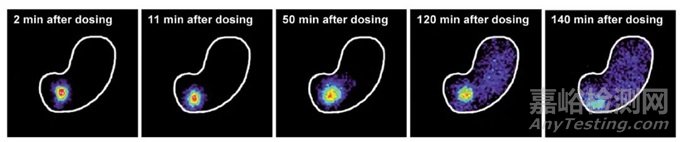
通過比較正常犬和幽門結扎犬口服索馬魯肽片劑后藥時曲線如圖6,幽門結扎后對索馬魯肽的吸收并沒有顯著的影響[9]。為了進一步證實索馬魯肽在胃部吸收,研究人員將犬脾靜脈底端和右側胃網膜靜脈結扎,此時脾靜脈血全部來自于胃部,而門靜脈血主要來自于小腸。口服索馬魯肽后分別采集脾靜脈血和肝門靜脈血。結果所示,脾靜脈中索馬魯肽的濃度遠高于肝門靜脈,說明索馬魯肽最初的攝取是發生在胃部,而不是小腸。
圖5:正常犬和幽門結扎犬口服索馬魯肽片劑后的濃度曲線
藥代動力學方面,圖7總結了口服索馬魯肽與皮下注射索馬魯肽的藥代動力學參數[6]。口服索馬魯肽采用連續用藥的方式(前5天5毫克/日,后5天10毫克/日,共10天),而后持續取樣21天,Cmax為15 nM,Tmax為1 h,半衰期為1周;皮下注射索馬魯肽單次用藥0.5毫克,Cmax為10 nM,Tmax為24 h,半衰期為1周。歸功于良好的藥代動力學性質,皮下注射索馬魯肽每周用藥一次即可,為糖尿病人提供了極大的便利。而口服索馬魯肽是目前唯一可以口服的GLP-1RA藥物,每日餐前半小時空腹服用一次即可,這無疑為糖尿病患者提供了更多的選擇。
索馬魯肽主要通過DPP-4蛋白及中性肽鏈內切酶的水解和脂肪酸二酸側鏈的氧化進行代謝,代謝產物通常通過糞便及尿液排出[10]。鑒于糖尿病病人常常需要聯合用藥,臨床上也考察了索馬魯肽對包括華法林和二甲雙胍等在內的多種常用藥暴露量的影響,并未觀察到索馬魯肽與之產生藥物-藥物相互作用[11,12]。
圖6:口服索馬魯肽與皮下注射索馬魯肽的藥代動力學參數
索馬魯肽的各項指標基本都優于或持平于其他GLP-1RA藥物,但其最亮眼的優勢還是在于給藥方式除了皮下注射以外還可以口服。值得一提的是,除了二型糖尿病,多項研究表明,索馬魯肽在減肥、降低Ⅱ型糖尿病患者重大心血管事件(Major Adverse Cardiovascular Events, MACE)風險、治療非酒精性脂肪性肝炎(Nonalcoholic Fatty Liver Disease, NASH)以及腎功能受益等方面也有積極影響。
在索馬魯肽及其口服制劑的研發過程中,主要關鍵點有兩個,一是如何提高藥物半衰期,二是如何實現口服。首先,為了解決多肽半衰期短的弊端,替換酶作用位點的氨基酸,以抵抗酶的水解;鏈接脂肪酸側鏈,以增強與白蛋白的結合,從而延緩系統清除,以達到長效的目的。為了實現口服用藥,使用一種小分子吸收增強劑,成功使口服索馬魯肽達到了足夠的生物利用度。SNAC可以局部提高胃內pH值,防止索馬魯肽被胃中的肽酶降解,同時促進索馬魯肽的吸收,突破了多肽口服不能吸收的限制。
參考文獻:
[1] 9th ed.Brussels: International DiabetesFederation, (2019).
[2] Cantini G, Mannucci E, Luconi M. Trends inEndocrinology & Metabolism, 2016, 27(6): 427-438.
[3] Knudsen L B, Lau J. Frontiers in endocrinology, 2019, 10:155.
[4] Kalra S,Sahay R. Diabetes Therapy, 2020: 1-18.
[5] Lau J,Bloch P, Scha?ffer L, et al. Journal of medicinal chemistry, 2015, 58(18):7370-7380.
[6] Pineo G, Hull R, Marder V.Best Practice & Research Clinical Haematology, 2004, 17(1): 153-160.
[7] Bittner B, McIntyre C,Tian H, et al. Die Pharmazie-An International Journal of PharmaceuticalSciences, 2012, 67(3): 233-241.
[8] Castelli M C, Wong D F,Friedman K, et al. Clinical therapeutics, 2011, 33(7): 934-945.
[9] Buckley S T, Bækdal T A,Vegge A, et al. Science translational medicine, 2018, 10(467).
[10] Bækdal T A, Thomsen M,Kup?ová V, et al. The Journal of Clinical Pharmacology, 2018, 58(10):1314-1323.
[11] PratleyR E, Aroda V R, Lingvay I, et al. The lancet Diabetes & endocrinology,2018, 6(4): 275-286.
[12] Gomez-PeraltaF, Abreu C. Drug design, development and therapy, 2019, 13: 731.