亞硝胺雜質一直是各大醫藥監管機構近些年來高度關注的話題,他們加大了對制藥公司和CDMO在亞硝胺檢測和風險評估方面的監管力度,導致了多個品種的藥品召回事件。
1、歐洲藥典委員會修訂涉及亞硝胺雜質的2034和2619通論
日前歐洲藥典 (Ph. Eur.) 委員會 (EPC, European Pharmacopoeia Commission) 在第 174 屆會議上通過了修訂后的通論 2034 和 2619,其中包括一段解釋 Ph. Eur對于亞硝胺雜質的控制方法。
通論2034的藥用物質Substances for pharmaceutical use,在“生產”下增加了一段關于 N-亞硝胺的內容:“由于許多 N-亞硝胺被歸類為可能的人類致癌物,因此負責制造人類藥物活性物質的制造商,需要評估在整個制造過程和儲存過程中 N-亞硝胺形成和污染的潛在風險。一旦風險得到確認,制造商應盡可能減少 N-亞硝胺的產生,例如通過修改制造工藝來實現,并應實施合理有效的控制策略,來檢測和控制這些雜質。制造商可以參照通用章節 2.5.42 :活性物質中的 N-亞硝胺。”
Pharmaceutical preparations (通論2619) 在亞硝胺的章節中增加了關于 N-亞硝胺的類似段落:“由于許多 N-亞硝胺被歸類為可能的人類致癌物,除了僅供獸醫使用的產品和未經許可的藥物制劑外,醫藥產品(medicinal products)制造商應評估在其整個生產過程和保質期中,N-亞硝胺形成和污染的潛在風險。如果風險得到確認,制造商應盡可能減少 N-亞硝胺的存在,例如通過修改制造工藝,并且必須實施控制策略來檢測和控制這些雜質。制造商可以參照通用章節 2.5.42 :活性物質中的 N-亞硝胺”。
2、歐洲藥典關于亞硝胺雜質分析的2.5.42通則
歐洲藥典委員會設定的關于亞硝胺雜質分析的2.5.42通則,應被視為一個分析工具箱,它提出了三種分析亞硝胺雜質的手段(GC-MS、LC-MS/MS 和 GC-MS/MS)。重要的是包括使用不同儀器,滿足歐洲及其他地區許多質量控制實驗室的不同需求。
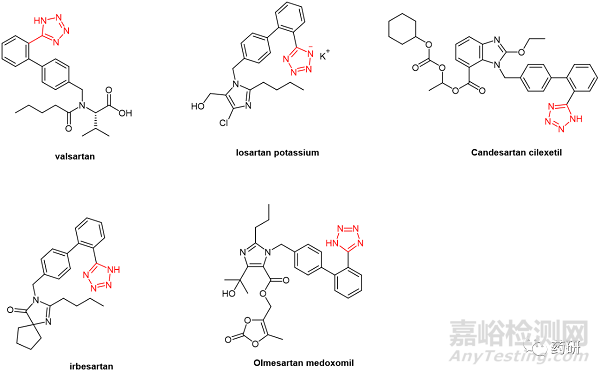
這些分析程序已針對所列亞硝胺物質進行了驗證,作為目標濃度為 30 ppb 的限度測試(limit test,程序 A 和 B),或作為定量測試(quantitative test,程序 C,GC-MS/MS)。該通則主要針對分析含有四唑基團的血管緊張素-II-受體拮抗劑(angiotensin-II-receptor antagonists,沙坦類藥物)中的 N-亞硝胺雜質,該類藥物有 5 個 Ph. Eur. 專著(纈沙坦、氯沙坦鉀、坎地沙坦酯、厄貝沙坦和奧美沙坦酯,圖1)。 除了這5種沙坦類藥物之外,其它的藥物可在額外的分析方法驗證之后,在證明預期目的的適用性條件下,通過以上的分析方法進行相關的亞硝胺雜質檢測。
圖1. 需要亞硝胺分析的五類沙坦類藥物分子結構
以上的三種分析方法針對了七種N-亞硝胺雜質(圖2):N-亞硝基二甲胺(NDMA)、N-亞硝基二乙胺(NDEA)、N-亞硝基二丁胺(NDBA)、N-亞硝基-N-甲基-4- 氨基丁酸 (NMBA)、N-亞硝基二異丙胺 (NDiPA)、N-亞硝基乙基異丙胺 (NEiPA) 和 N-亞硝基二丙胺 (NDPA)。
圖2. 歐洲藥典規定的7種受檢亞硝酸雜質
為支持新通過的關于活性物質中 N-亞硝胺雜質分析的通則(2.5.42)的實施,已經建立了圖2中描述的七種參考標準品,可從歐洲理事會藥品和醫療保健質量 (EDQM,European Directorate for the Quality of Medicines & HealthCare) 獲取。
歐洲藥典委員會設定的關于亞硝胺雜質分析的2.5.42通則,是在幾個官方藥物控制實驗室 (OMCL) 的幫助下創建的。該通則并未提供用于測試 N-亞硝胺雜質的分析程序的詳盡清單。根據亞硝胺雜質分析的困難程度以及測試需求,其它的分析手段可能會更適合,但需要經過分析手段的驗證過程。
鑒于亞硝胺雜質在全球使用的藥物中被廣泛發現,Ph. Eur和 USP 將致力于趨同,以確保其公共質量標準保持一致。