藥物的多晶型即指藥物存在兩種或以上不同晶型物質,可能包括水合物晶型、無水合物晶型、溶劑合物晶型等。不同晶型可能會對藥物的理化性質產生較大影響,比如晶型粒子的表面積、結晶狀態和粒徑大小會影響藥物的溶出速度;不同結晶粒子還會影響藥物的溶解度、生物利用度和穩定性等,從而影響產品的有效性、安全性及質量可控性。但有些時候,企業也會選擇這些高能量、亞穩態晶型或者無定形來提高產品的生物利用度或者規避有關專利等,故對于多晶型藥物,國際藥物監管機構會要求新藥和仿制藥的申報者提供資料充分證明對其晶型有足夠了解和控制,需要企業建立晶型藥物的質量標準,包括晶型種類、純度、比例以及質量控制標準,對于晶型的質量控制,則需要開發一種可用于晶型定性、定量檢測分析的方法,來保證藥物質量的一致性、穩定性、安全性及有效性。
關于不同晶型物質的鑒別,常用的定性檢測方法有:光學顯微鏡法、偏光顯微鏡法、單晶X射線衍射法(SXRD)、粉末X射線衍射法(PXRD)、拉曼光譜法、差示掃描量熱法(DSC)、紅外光譜法(IR)、動態水吸附法(DVS)、熱重法(TGA)等。晶型定量檢測方法可能包括有:單晶X射線衍射法、粉末X射線衍射法、紅外光譜法、差示掃描量熱法等,本文就這幾種常用的定量方法做簡單介紹。
1. X射線衍射法
X射線衍射法是利用原子對X-射線的衍射效應,完成對物質結構、成分、物質晶型的定性、定量研究。它是國際公認的晶型藥物定量質量控制的重要手段。它包括單晶X射線衍射法(SXRD)和粉末X射線衍射法(PXRD)。單晶X射線衍射法是以一顆單晶體作為研究對象,從分子層面給出不同晶型藥物的分子排列規律、分子構象結構等三維立體結構信息、結晶水含量等定量信息;粉末X射線衍射法則是以無數粉晶物質樣品作為研究對象,可用于物質狀態、成分、晶型質量控制等分析研究。其中,粉末X射線衍射法是常用的分析檢測方法,它的圖譜如同人的指紋一樣,由衍射峰數量、位置、峰強、衍射峰幾何拓圖特征構成。當晶型固體物質是由兩種或以上的混合晶型物質組成時,其粉末X射線衍射圖譜是按照每種晶型物質特征性圖譜進行物理方式的疊加,混晶樣品的衍射圖譜衍射峰強度也會隨著樣品混合比例的改變而變化。該方法使用方便,被測樣品不需要復雜的前處理過程,僅需研磨即可,可適用于原料藥的晶型分析、制劑中的晶型種類及不同晶型的定量分析。PXRD的定量分析方法又包括單峰法和全譜擬合法[1]。
單峰法:即選取特征衍射峰,建立特征衍射峰峰強度與被測藥物中各晶型組分的對應關系。同一藥物在不同混晶的情況下,建立混合物中的各種晶型含量與特征峰衍射強度關系的標準曲線,可以實現對原料藥的晶型種類和比例的含量測定。該方法簡單,對樣品的信息要求較低,靈敏度也相對較高,是常用的一種定量分析方法。例如有文獻應用PXRD的單峰法測定地氯雷他定中晶型Ⅰ和晶型Ⅱ的比例,首先選擇不同晶型的特征衍射峰,晶型Ⅰ選擇2θ=29.4°為特征衍射峰,晶型Ⅱ選擇2θ=22.4°為特征峰,以5份晶型Ⅰ和晶型Ⅱ的不同比例混合對照品樣品的特征峰面積和重量比進行線性回歸,得到線性方程:y=2.3205x-1.5641(r=0.9986)。


應用建立好的標曲對實際不同批次的樣品進行測定,計算晶型Ⅰ的含量,由結果可知樣品中晶型Ⅱ的比例較小,基本不超過10%[2]。
單峰法雖簡單,但是它也有弊端之處,比如單峰法的定量方法嚴重依賴于標樣的純度,而且制備樣品時所產生的擇優取向效應也會影響該方法的準確性;且由于樣品受到輔料的稀釋,樣品的特征峰強度會被削弱,所要求的檢測限提高等。因此,晶型的單峰定量方法在實際應用中需要嚴格控制操作條件,且通常也會與其他方法例如XRPD會和傅里葉紅外光譜或拉曼光譜聯合,以減少相應誤差。
全譜擬合法:即建立全譜峰強度與被測藥物中各晶型組分的對應關系。它是以化學計量學為基礎,如偏最小二乘法、全粉末圖譜分解法、Rietveld法等方法對樣品進行定量分析,其信噪比、靈敏度和專屬性均比單峰法有明顯提高。該方法不需要提供不同晶型樣品的標樣,但是對樣品信息的要求較高,需要提供混合物中每個相的晶格參數、空間群對稱性、質量吸收系數、原子坐標、位置占有率等參數來模擬粉末衍射圖譜,雖定量測定結果更加準確,但是方法建立和計算過程非常復雜,對樣品圖譜的解析能力要求也較高,所以實際推廣可能應用較少。如有文獻通過Rietveld法使用Fullprof軟件建立了法莫替丁晶型A和晶型B混合物的定量分析方法,該方法首先是需要結合XRPD圖譜得到一個計算模型,然后用含量為50%(w)的晶型A和晶型B的混合物對該計算模型進行修正,修正后即可用于晶型的定量分析,由結果可知計算值與實際值非常貼合,準確度很高[3]。
2. 紅外光譜技術
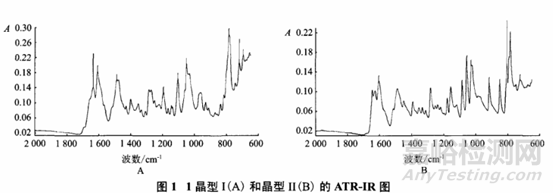
不同晶型藥物的紅外吸收光譜差異,主要表現在峰形變化、峰位偏移、峰強度改變等。在用紅外照射時,分子中的化學鍵或官能團可發生振動吸收,不同的化學鍵或官能團吸收頻率不同,因此在紅外光譜上將處于不同位置,從而可以獲得分子中任何化學鍵或官能團的信息。但對于多晶型樣品,由于不同晶型樣品的分子結構完全相同,因而只有在不同晶型分子間作用力發生變化或者溶劑分子介入時,才能表現出圖譜的差異,且該方法易受樣品中雜質的干擾影響,其譜圖差異不及XRD圖譜差異明顯,因此一般只作為定性鑒別和輔助定量鑒別方法。在藥物的多晶型分析過程中,由于樣品在研磨過程中可能會造成藥物的晶型發生變化,因此常用石蠟糊法或擴散反射傅里葉紅外光譜法進行紅外光譜測定。例如有研究者采用衰減全反射紅外光譜法測定拉米夫定晶型Ⅱ的含量,先選擇晶型Ⅱ的定量吸收峰,由圖譜可知晶型Ⅱ在850cm-1處有明顯的吸收峰,而晶型Ⅰ在此處無干擾,因此選定該位置作為晶型Ⅱ的特征吸收峰。
通過配置不同比例的晶型Ⅰ和晶型Ⅱ混合物樣品,將圖譜進行疊加,并在500~1000cm-1范圍內進行滿刻度顯示,由圖知785cm-1的吸收峰為最大峰,850cm-1的吸收峰強度呈明顯的濃度依賴性,因此把785cm-1的吸收峰作為內標峰,以定量峰和內標峰的吸收度之比對濃度進行線性回歸,得到線性方程A850/A785=5.949*10-3C+0.00902(R=0.9935)。對不同批的原料或片劑進行紅外光譜測定,可知:原料的晶型純度均較高;而制劑樣品中有一批樣品中晶型Ⅱ的含量為93.7%[4]。
3. 差示掃描量熱法(DSC)
該方法是基于熱力學原理和物質熱力學性質而建立的分析方法。利用不同晶型物質特有的熱力學性質,通過供試品吸熱峰或放熱峰的數量、位置、形狀、吸熱量等參數變化實現對晶型物質狀態的定性、定量鑒別。該方法操作簡單,試樣用量少且樣品不需要預處理,但它僅適用于不同晶型物質的熔融吸熱峰存在較大差異或供試品中含有不同數量和種類結晶溶劑的晶型物質的鑒別。例如有文獻應用差示掃描量熱法定量研究阿德福韋的不同晶型,分別對A、E兩種晶型樣品進行DSC分析,結果可知兩種晶型具有明顯不同的熔點,A晶型的熔點為100℃,E晶型的熔點為93℃,兩種晶型的熔融峰可以明顯區分開,有利于利用熔融熱焓的數據對不同晶型的含量進行定量分析。分別配置含不用比例的A和E晶型的混合樣品,進行DSC測定,混合物樣品測量用量約1mg左右,混合物DSC的測試曲線結果如圖5。
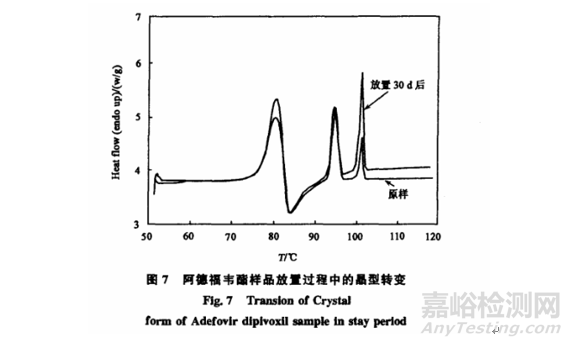
應用該DSC方法測定阿德福韋樣品在二氯甲烷溶劑中0~5℃重結晶獲得的樣品以及在室溫20~25℃放置30d后的樣品,通過計算可以知道樣品中A晶型的含量從11.2%提高到了25.6%,即在室溫放置過程中有部分非晶組分轉成了A晶型[5]。
4. 拉曼光譜法
拉曼光譜法是以拉曼效應為基礎研究分子振動的一種方法,是研究分子和光相互作用發生散射光的頻率,該方法可以利用不同晶型物質特有的分子極化率變化,引起指定波長范圍的拉曼光譜吸收峰位置、強度、峰形幾何拓撲等參數變化,從而實現對晶型的定性、定量鑒別。而同種化合物的不同晶型,由于其堆積方式不同會導致分子振動有所不同,故而可以用來區分多晶型物質。拉曼光譜法和紅外光譜法均屬于振動光譜,但拉曼光譜檢測低頻振動較紅外光譜具有更明顯的優勢,甚至可以檢測到分子的晶格振動,因此拉曼光譜在鑒別藥物的多晶型方面具有顯著的優勢作用。該方法測定簡單,檢測樣品不需要預處理,可直接檢測,可以避免樣品處理過程中對晶型的影響。但該方法尚不成熟,暫不具權威性,一般可作為晶型定量分析的輔助分析手段。熒光干擾是拉曼光譜分析中常遇到的問題,需要通過摸索合適的曝光時間和采樣次數,并且采用基線矯正,可以一定程度上減少熒光干擾。另外拉曼光譜信號絕對強度波動較大,使用同一樣品多次測量,衍射峰強度的偏差可能達到20%,因此常采用內標法來定量分析,固體樣品可以采用樣品中某一位置的峰強作為內標或參比,可以一定程度上提高定量結果的準確性[6]。
例如有研究者應用拉曼光譜法輔助定量檢測拉米夫定晶型Ⅰ的含量。研究者先用XRD定量分析Ⅰ晶型,發現雖然Ⅰ晶型的特征峰明顯,但是峰強明顯弱于晶型Ⅱ,且晶型Ⅰ為針狀晶體,擇優取向會導致特征峰的強度重現性較差,較難應用于晶型Ⅰ的定量分析,而在拉曼光譜中,晶型Ⅰ在697cm-1有明顯的特征散射峰,晶型Ⅱ在該位置處無干擾,因此用拉曼方法輔助定量分析晶型Ⅰ的含量。將不同比例的晶型Ⅰ和晶型Ⅱ混合物樣品的圖譜進行疊加,發現697cm-1 散射峰強度呈明顯的濃度依賴性,選擇該位置作為定量峰;兩種晶型在537cm-1處均有散射峰,且該位置未受其他峰干擾,峰形良好,將該位置作為內標峰,采用定量峰和內標峰的強度比對濃度進行線性回歸,得到線性方程為I697/I537=7.152*10-3C+0.003143(R=0.9986)。取原料適量進行拉曼光譜測定,采用標準曲線法進行計算,由圖5結果可知并未檢出晶型Ⅰ[6]。
另外除了以上幾種定量方法外,如果原料的不同晶型物質溶解性質存在較大差異時,也可以嘗試建立晶型物質與溶解度或溶出度的關系研究,以溶解度或者體外溶出度、溶出速率作為晶型的定量評價輔助方法。本身藥物所存在的多晶型研究就比較復雜,還要做到準確的定量研究就更難,我們一般會結合各個晶型的特點比較多種表征方法,最終選擇特征性更強、重現性更好的檢測方法。如果有可能的話,通常還會借助多種檢測技術的聯合應用來提高晶型定量檢測的準確性。
參考文獻:
[1] 馬樂偉等,藥物晶型定量分析方法的研究進展[J].藥學學報,2011,46(8):896-903
[2] 鄒文博等,應用粉末X射線衍射法測定地氯雷他定中晶型Ⅰ和晶型Ⅱ的比例[J].中國藥事,2018,32(5):637-641
[3] 肖燕等,固體藥物晶型定量分析方法[J].石油化工,2015,44(1):11-17
[4] 葉曉霞等,衰減全反射紅外光譜法測定拉米夫定晶型Ⅱ的含量[J].中國醫藥工業雜志,2009,40(12):928-930
[5] 袁鉆如等,差示掃描量熱法(DSC)定量測試阿德福韋晶型的研究[J].分析測試技術與儀器,1008,14(2):105-108
[6] 葉曉霞等,拉曼光譜法檢測拉米夫定晶型Ⅰ[J].中國醫藥工業雜志,2013,44(1):60-63。