您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2024-08-12 08:50
對于境外已上市境內未上市的藥品,需結合藥品具體情況,按照臨床評價的基本邏輯對原研藥品的臨床研究數據進行充分評價,根據評價結果確定臨床試驗路徑。對于不同研發背景的藥品,其所需開展的臨床試驗應具體問題具體分析。
1、法規要求
根據化學藥品注冊分類改革工作方案(2016年51號),化藥3類指境內申請人仿制境外上市但境內未上市原研藥品的藥品。該類藥品應與原研藥品的質量和療效一致。
原研藥品指境內外首個獲準上市,且具有完整和充分的安全性、有效性數據作為上市依據的藥品。
根據《境外已上市境內未上市藥品臨床技術要求》(2020年29號),境外已上市境內未上市藥品的臨床技術要求,應遵循臨床評價基本邏輯,在充分評價中國患者臨床需求、境外原研藥品臨床安全性和有效性、以及種族因素影響的基礎上,基于中國患者獲益/風險評估的需要,確定其在境內上市需開展的臨床試驗技術要求。
臨床評價基本邏輯
(一)臨床需求評估
對中國患者臨床需求的程度做出判斷。
(二)有效性和安全性評價
(三)種族敏感性分析
(四)基于中國患者獲益/風險評估進行決策
CDE培訓會議中也講到通常豁免臨床是比較難的,通常也只是限于嚴重危及生命及罕見病等的一些優先審評品種。

2、案例分析
根據實際情況化藥3類又可以分為真3類(全新的化合物進入國內)、半真3類(國內有其它劑型產品)、假3類(國內有同品種仿制藥上市多年),不同情況所進行的臨床試驗也不同。
3類化藥可能需要進行的臨床試驗情形如下:
第一種是豁免BE及其他臨床試驗,由境內外臨床數據支持,通過體外評價證實與原研一致;
第二種是BE試驗,可豁免驗證性臨床試驗僅以BE試驗申請注冊;
第三種是驗證性臨床試驗,無法或無需進行BE試驗的品種,通過驗證性臨床試驗證實有效及安全性;
第四種是BE+驗證性臨床試驗,BE證實與原研生物等效,驗證性臨床確證療效及安全性;
第五種是劑量探索(I期)+確證性臨床研究,有種族差異的,需以新藥臨床思路進行仿制藥臨床研究。
根據BE試驗中的PK數據,可以結合原研公布的PK數據進行對比,多角度分析其是否具有PK方面的種族差異,若無PK差異,則可考慮直接開展驗證性臨床;若有種族差異,按照臨床評價的基本邏輯則還應考察中國人群的劑量探索性試驗,為驗證性臨床提供劑量依據。
(1)BE+驗證性臨床
做了驗證性臨床的3類仿制藥絕大多數都是國內首次獲批的化合物,如奧貝膽酸片,江蘇恒瑞只做了BE進行申報未被批準;正大天晴做完BE 后,后面申請了3期臨床。

氟比洛芬酯注射液(特殊注射液),該品種雖然有2家仿制藥上市,但原研未進入國內,仍按3類申報,武漢大安進行了3期臨床、1期臨床和PK試驗;廣東嘉博制藥有限公司2018年完成BE試驗后進行申報未被批準,2020年完成III期臨床后進行申報,目前已經獲批。四川科倫、山東威高、揚子江藥業等沒做臨床的企業全部被斃掉,根據審評結果可以發現,仿制該品種仍然需要進行有效性和安全性的臨床試驗:像氟比洛芬酯這種情況的項目還有很多品種。
同類藥物CDE臨床登記情況如下:
|
|
藥物名稱 |
臨床試驗類型 |
|
1. |
替米沙坦氨氯地平片 |
參比制劑原研已經進口,生物等效性試驗 |
|
2. |
奧美沙坦酯氨氯地平片 |
參比制劑原研地產化品種,生物等效性試驗 |
|
3. |
纈沙坦氨氯地平片(Ⅱ) |
參比制劑原研已經進口,生物等效性試驗 |
|
4. |
阿齊沙坦氨氯地平片 |
參比制劑未進口原研藥品,日本橙皮書,CDE臨床登記情況如下,BE+驗證性臨床
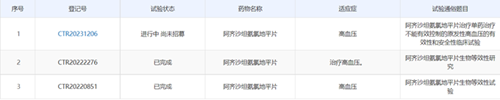  |
|
5 |
厄貝沙坦氨氯地平片 |
參比制劑未進口原研,歐盟、日本上市。
根據CDE臨床登記為BE加驗證性臨床試驗
|
(2)生物等效性試驗(BE)
1)僅進行BE獲批的品種中,此類品種多為緩控釋制劑,例如鹽酸美金剛緩釋膠囊、苯扎貝特緩釋片等,由于其普通片劑已在國內上市多年,因此僅需BE證實與原研生物等效,豁免驗證性臨床試驗。

特例,拉考沙胺注射液國內首仿企豁免臨床的關鍵在于該藥的藥代特性為口服生物利用度約為100%,原研片劑批準進口,有國內臨床數據充分,注射劑通過與片劑BE等效豁免臨床。
(3)豁免臨床
廠家沒做驗證性臨床也獲批上市的注射劑產品有:鹽酸溴己新注射液、克林霉素磷酸酯注射液、硫酸特布他林注射液、左西孟旦注射液和注射用右雷佐生等。這些不需要做臨床的產品都有一個特點,就是仿制藥廠家已經在國內上市多年的注射劑。
原研小針及國產粉針、小針上市多年,療效安全性充分驗證;100ml:4mg唑來膦酸注射液的原研產品明確,與已上市品種適應癥、用法用量相同,最終獲準豁免驗證性臨床試驗。
3、臨床試驗技術要求
已廢止的舊版《藥品注冊管理辦法》2007年28號以及對應的附件2:化學藥品注冊分類及申報資料要求
“3.已在國外上市銷售但尚未在國內上市銷售的藥品:(1)已在國外上市銷售的制劑及其原料藥,和/或改變該制劑的劑型,但不改變給藥途徑的制劑;”
“2.屬注冊分類3和4的,應當進行人體藥代動力學研究和至少100對隨機對照臨床試驗。多個適應癥的,每個主要適應癥的病例數不少于60對。避孕藥應當進行人體藥代動力學研究和至少500例12個月經周期的開放試驗。
屬于下列二種情況的,可以免予進行人體藥代動力學研究:
(1)局部用藥,且僅發揮局部治療作用的制劑;
(2)不吸收的口服制劑。”
這是之前3.1類(即現在的新3類)需要做臨床的法規依據。
現行《藥品注冊管理辦法》2020年27號以及對應的NMPA 化學藥品注冊分類及申報資料要求 20200629
“3類:境內申請人仿制境外上市但境內未上市原研藥品的藥品。該類藥品應與參比制劑的質量和療效一致。”屬于之前3.1類,但是去掉了明確的臨床試驗要求。而是在“管理辦法”正文中:
“第三十五條 仿制藥、按照藥品管理的體外診斷試劑以及其他符合條件的情形,經申請人評估,認為無需或者不能開展藥物臨床試驗,符合豁免藥物臨床試驗條件的,申請人可以直接提出藥品上市許可申請。豁免藥物臨床試驗的技術指導原則和有關具體要求,由藥品審評中心制定公布。”后面CDE出臺了《境外已上市境內未上市藥品臨床技術要求》(2020年29號)
新的藥品注冊管理辦法對3類藥取消了“PK+100例”隨機對照臨床試驗的規定,制定出以科學為基礎、同時基于ICH E5、E17、E6技術要求,更加靈活科學的臨床試驗方案。新3類取消了監測期,對首仿和后續申報申請人的臨床研究要求尚未在公開的規定上予以明確,3類藥開展驗證性臨床試驗存在一定的顧慮。
CDE后續是否可行考慮首家獲批后給予市場獨占期,之后申報者可以BE或者直接報產,這樣給首家帶來好處的同時也避免了資源浪費,無需重復做臨床試驗。
此外,從網絡各個渠道能得到的CDE審評人員對于該問題的回復,基本上是“企業需要自行評估,case by case”以及“基于充分的數據與分析,與CDE提前溝通。
通過CDE一般性技術問題咨詢得到的答復也是建議申請人明確具體品種提交溝通交流申請。


來源:注冊圈



