您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2021-11-30 12:40
在進行藥物的有關物質檢查時,多采用高效液相色譜法,其計算方法有外標法、加校正因子的自身對照法、自身對照法及峰面積歸一化法。其中加校正因子的自身對照法定量準確,又無需在每次檢測時均提供雜質對照品,是目前較為常用的一種方法。小編在此對校正因子的測定以及校正因子準確度的驗證進行簡述。
一、校正因子的測定
校正因子的測定有單點法、多點法、標準曲線法及吸收系數比值法。已有多篇文獻報道過各方法的基本操作及優缺點,在此不再贅述。在進行校正因子測定時,小編建議采用標準曲線法,因為線性試驗是方法驗證必不可少的一項內容,在此采用標準曲線法,既驗證了雜質對照品及主成分的線性,又可得到準確的校正因子,可謂一舉兩得。
在進行校正因子的測定時,首先將各雜質對照品及主成分對照品配制成一定濃度的儲備液,逐步稀釋,直至找到各雜質的定量限,線性范圍應包括定量限濃度~限度濃度的150%,主成分的濃度范圍應包括定量限濃度~自身對照濃度的150%。例如,有關物質測定時,供試品的濃度為1mg/ml,采用0.1%的自身對照(即自身對照的濃度為1μg/ml),雜質A的限度為0.1%(即雜質A的限度濃度為1μg/ml),假設主成分及雜質A的定量限均為20ng/ml,則雜質A和主成分應在20ng/ml~1.5μg/ml的范圍將濃度與峰面積進行線性回歸,相關系數應大于0.990,Y軸截距應在100%響應值的25%以內。繪制出各雜質及主成分的線性曲線后,各雜質與主成分線性方程斜率的比值即為該雜質的校正因子。若雜質的校正因子在0.9~1.1的范圍內,無需驗證,直接采用自身對照法,若雜質的校正因子0.2~5.0之間,則需要采用加校正因子的自身對照法,若雜質的校正因子小于0.2或大于5.0,可考慮變換雜質的檢測波長,重新進行測定,若所有的測定均在0.2~5.0之外,則表示加校正因子的自身的對照法不適用,需要用外標法進行定量。
在配制各濃度的線性溶液時,一定注意需逐級稀釋,要避免出現下圖1所示的情況,應如圖2所示,保證個點分布的均勻性。
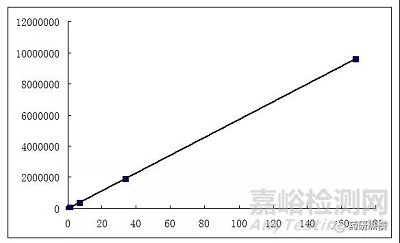
圖1 不正確的線性試驗設計

圖2 正確的線性試驗設計
二、相對校正因子及相對保留時間耐用性的考察
在采用加校正因子的自身對照法進行定量時,一般采用相對保留時間對雜質進行定位,在此,一并對相對校正因子及相對保留時間的耐用性進行考察。
具體考察方法同其他方法驗證的耐用性試驗,即變換波長、流速、柱溫、流動相比例、流動相pH值、采用不同品牌的色譜柱、儀器等。將各雜質對照品與主成分配制成混合對照品溶液,采用標準曲線法,繪制各條件下雜質對照品及主成分對照品的線性方程,計算校正因子,同時計算出各條件下雜質對照品相對于主成分的相對保留時間。
對相對校正因子,除波長的變化會對相對校正因子有所影響外,其余色譜條件的調整均不會對相對校正因子產生影響,因此,測定波長的選擇要避開吸光度急劇變化的位置,應選擇在最大吸收或最小吸收的位置。校正因子的變化范圍在多少范圍內可以接受,目前,沒有定論,小編認為,如果最終標準中相對校正因子只保留到小數點后一位,例如0.7,則各條件下校正因子均控制在0.65~0.74范圍內,即可判定耐用性較好。即采用有效數字的修約規則,修約后為標準中規定的數值即可。
對相對保留時間,除波長的變化對其沒有影響外,其余各條件的變化均會對其產生影響,為了保證對雜質的準確定位,必須對相對保留時間的耐用性進行考察。此處的試驗不是為了證明相對保留時間的耐用性良好,而是通過耐用性考察確定出如何采用相對保留時間對雜質進行定位。例如,固定色譜柱的填料及品牌,固定主峰的保留時間等。至于RRT在多少范圍內可以接受,不能一概而論,若雜質譜較復雜,則質量標準中的RRT需要保留至小數點后兩位,如果雜質譜相對簡單,各雜質距離較遠,RRT保留至小數點后一位即可。在此,跟大家分享另外一個經驗,即如果某些雜質的相對保留時間無法固定,隨色譜參數的變化較明顯,則可以采用將主成分進行定向破壞,通過系統適用性試驗來對雜質進行定位。
三、相對校正因子準確度的考察
配制相同的供試品溶液,同時采用加校正因子的自身對照法及外標法,對供試品中的雜質含量進行測定,對兩種方法的測定結果進行比較,校正因子與外標法計算結果的負偏差不得過0.05%,正偏差不做規定。若供試品中檢出的雜質較少,可在供試品中加入一定量的雜質對照品,來進行測定。
至此,可將相對校正因子固化到質量標準中,采用加校正因子的自身對照法對已知雜質進行定量計算,既保證了定量結果的準確,又簡化了試驗,節省了對照品的用量。

來源:Internet


