您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2020-09-22 16:25
1、什么是除菌過濾,什么是除菌過濾器?
依據國家藥監局2018.10.1生效版《除菌過濾技術及應用指南》中的定義:除菌過濾是指采用物理截留的方法去除液體中的微生物,以達到無菌藥品相關質量要求的過程。
除菌級過濾器指在工藝條件下每平方厘米有效過濾面積可以截留大于等于1×107cfu的缺陷型短波胞菌(Brevundimonasdiminuta,曾用名:缺陷假單胞菌)的過濾器。具體可參考ASTM 838-15,使用缺陷型短波胞菌對除菌級過濾器進行挑戰,以確認除菌過濾器的微生物截留能力。
2、除菌過濾器產品為什么要做驗證?
主要基于藥品(產品)與過濾器之間的相互影響,有來自于產品潤濕后的完整性試驗,以及過濾器對產品的溶出物(可提取物或浸出物)試驗,吸附試驗,還有產品對過濾器細菌截留率方面的影響。這些影響對應需要做相關的試驗來驗證。
3除菌過濾器可參考的法規有哪些?
除菌過濾器可參考的相關驗證的法規如下:
US FDA 21 CFRPART 211.65
FDA Guideline onSterile Drug Products Produced by Aseptic Processing Sep. 2004
EU GoodManufacturing Practices (2016 draft) and the Annex 1: Manufacture of Sterile Products (2020 draft)
國家藥品監督管理局GMP無菌藥品/無菌藥品附錄 (2010 版)
InternationalStandard ISO 13408-2 Part 2: Filtration (2018)
PDA TechnicalReport No.26-Sterilizing Filtration of Liquids, Revised 2008
滅菌無菌工藝驗證指導原則sterile, aseptic process validation guideline
國家藥品監督管理局《除菌過濾技術及應用指南》(2018.10)
《化學藥品注射劑滅菌和無菌工藝研究及驗證指導原則》(2020.08)
4、除菌過濾器驗證內容有哪些?
依據國家藥監局2018.10.1生效版《除菌過濾技術及應用指南》中5.1除菌過濾驗證概述,表明除菌過濾器驗證內容包括:性能確認(由過濾器廠商-依據藥典方法進行)和過濾工藝驗證(由過濾器用戶或委托第三方實驗室經評估來設計試驗)兩部分。下圖展示出性能確認和過濾工藝驗證對應的項目。
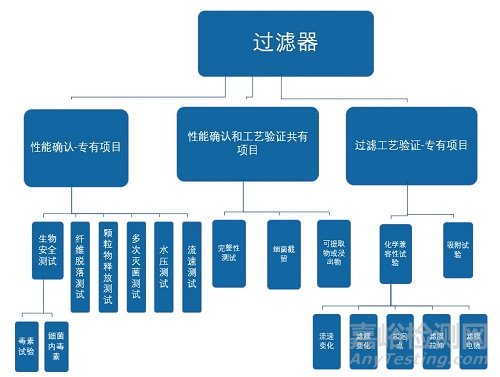
5、除菌過濾器的過濾工藝驗證具體有哪些?
依據國家藥監局2018.10.1生效版《除菌過濾技術及應用指南》中5.1除菌過濾驗證概述,過濾工藝驗證是指針對具體的待過濾介質,結合特定的工藝條件而實施的驗證過程,一般包括細菌截留試驗、化學兼容性試驗、可提取物或浸出物試驗、安全性評估和吸附評估等內容。如果過濾后,以產品作為潤濕介質進行完整性測試,還應進行相關的產品完整性測試驗證。除菌過濾工藝驗證可以由過濾器的使用者或委托試驗檢測機構(例如:過濾器的生產者或第三方試驗室)完成,但過濾器使用者應最終保證實際生產過程中操作參數和允許的極值在驗證時已被覆蓋,并有相應證明文件。
一、完整性測試:應明確過濾器使用后完整性測試的潤濕介質(包括誰、乙醇等標準介質或藥液)。如果采用的潤濕介質為藥液,則應進行產品相關完整性標準的驗證以支持該標準的確定。完整性測試貫穿于細菌截留試驗以及化學兼容性試驗,除此之外,在藥品連續三批生產工藝驗證及常規生產過程中也涉及完整性測試。(來源于化學藥品注射劑滅菌滅菌/無菌工藝研究及驗證指導原則。)
二、細菌截留:細菌挑戰試驗的研究目的是模擬實際生產過濾工藝中的最差條件,過濾含有一定量挑戰微生物(根據ASTM 838-15,使用缺陷型短波胞菌(Brevundimonas diminuta,曾用名:缺陷假單胞菌)對除菌級過濾器進行挑戰的產品溶液或者產品替代溶液,以確認除菌過濾器的微生物截留能力。(來源于國家藥監局2018.10.1生效版《除菌過濾技術及應用指南》章節5.2)。
三、可提取物或浸出物:可提取物試驗在選擇模型溶劑之前必須對產品(藥品)處方進行全面的評估。用于測試的模型溶劑應能夠模擬實際的藥品處方,同時與過濾器不應有化學兼容性方面的問題。可以從過濾器及其他組件材料的工藝介質接觸表面提取出的化學物質。應先獲得最差條件下的可提取物數據,將其用于藥品的安全性評估。可提取物反映了浸出物的最大可能。除菌過濾器的浸出物研究,需要根據提取研究結果和風險評估結果確定。一般可提取物和浸出物試驗的研究策略,是在可提取物評估基礎上,針對高風險的組件進行進一步的浸出物研究。應針對過濾器可提取物或浸出物的種類和含量,結合藥品最終劑型中的濃度、劑量大小、給藥時間、給藥途徑等對結果進行安全性評估(參考化學藥品注射劑生產所用的塑料組件系統相容性研究技術指南,一般使用SCT/TTC/QT/PDE進行評估,以評估可提取物和浸出物是否存在安全性風險。(來源于國家藥監局2018.10.1生效版《除菌過濾技術及應用指南》章節5.3)。
四、化學兼容性試驗:過濾器化學兼容性說明該工藝流體及其工藝條件對該過濾器的特定性能沒有產生負面影響。通常, 過濾器的性能包括過濾器的物理強度(在一定溫度下, 能耐受一定的壓差)、允許流體通過的能力(即通透性,如流速和通量)和去除流體中粒子的能力(即截留性能,如細菌、病毒或顆粒)。
化學兼容性試驗檢測項目一般有:過濾器接觸待過濾介質前后的目視檢查;過濾過程中流速變化;濾膜重量/厚度的變化;過濾前后起泡點等完整性測試數值的變化;濾膜拉伸強度的變化;濾膜電鏡掃描確認等。
PDA 26號第4.1章節:提到完整性測試和細菌挑戰測試的結果可以用來評估化學兼容性。
五、吸附試驗:吸附是所過濾的料液中的某些成分粘附在濾膜上的過程,可能影響料液的構成和濃度。過濾器中吸附性的材料包括濾膜、硬件和支撐性材料。吸附試驗條件可以根據實際生產條件確定,流速、過濾時間、料液濃度、防腐劑濃度、溫度和pH 值等因素都可能影響吸附效果。吸附試驗中采用的檢測方法可以采用產品質量標準中所確定的相關檢測方法。(來源滅菌無菌工藝驗證指導原則sterile,aseptic process validation guideline,3.2.2.4吸附試驗)。

來源:NSF認證檢測


