您當前的位置:檢測資訊 > 科研開發(fā)
嘉峪檢測網(wǎng) 2025-07-27 09:23
本技術報告(Technical Report,TR)旨在為制藥工藝驗證(PV)生命周期方法的實施提供實用指南。它包含的信息能夠使生產(chǎn)廠家實施全球認可的工藝驗證程序,該程序與最近的基于生命周期的工藝驗證指導文件和現(xiàn)行對藥品質(zhì)量系統(tǒng)期望相一致。在醫(yī)藥生產(chǎn)中,“工藝驗證”是收集和評價工藝設計階段的數(shù)據(jù),通過商業(yè)化生產(chǎn)的方式確定科學的證據(jù),證明一個工藝能夠持續(xù)地提供高質(zhì)量的產(chǎn)品。美國 FDA 和歐洲藥監(jiān)局認定 PV 在現(xiàn)行 GMP 指南的綜述和具體條框中都是必要條件和藥品質(zhì)量保證的必需元素。
工藝驗證生命周期概念連接產(chǎn)品和工藝開發(fā)、商業(yè)化生產(chǎn)確認和協(xié)同努力下商業(yè)化生產(chǎn)過程的維持。當基于良好的工藝理解和使用質(zhì)量風險管理時,生命周期法可考慮生產(chǎn)者在使用傳統(tǒng)的工藝驗證外,再使用連續(xù)工藝確證(增強的方式),或者直接由后者代替前者。
在本技術報告中應用于藥物和藥品生產(chǎn)過程的信息,包括:
無菌和非無菌藥物
生物技術/生物產(chǎn)品,包含疫苗
原料藥(APIs)
放射性藥物
獸藥
組合產(chǎn)品的藥物成分(如,復方抗菌藥和醫(yī)療器械)
這份報告是為全球使用和應用到新的和現(xiàn)有的(即遺留的)商業(yè)生產(chǎn)過程做準備。它的范圍不包括:
生產(chǎn)的生產(chǎn)過程如下:醫(yī)療器械,膳食補充劑,藥用物料,人體組織。
盡管這些產(chǎn)品分類超出了這份 TR 的范圍,但它的建議是基于現(xiàn)代質(zhì)量思想的/ICH質(zhì)量指南和最近的監(jiān)管機構權威指導性文件。因此,它可能在其他產(chǎn)品類別的工藝驗證生命周期法的開發(fā)中是有用的參考文獻。藥物生產(chǎn)過程中的輔助操作的驗證在這個報告中不做討論。一些 FDA 的技術報告已經(jīng)提供了此類操作規(guī)程的具體指導性文件;如:清潔、無菌過程模擬、濕熱滅菌和干熱滅菌。
工藝知識的建立和獲取(第一步-工藝設計)
本節(jié)集中于開發(fā)過程中所用的方法以實現(xiàn)具耐受性的生產(chǎn)工藝,它解決了工藝驗證第一階段中工藝和產(chǎn)品知識被開發(fā)以建立控制策略。風險評估和管理被使用以集中于研發(fā)工作。工藝和產(chǎn)品知識通過藥物研發(fā)方案逐步形成。為對工藝驗證用生命周期方法而設計一個綜合的、有效的程序/方案強迫在研發(fā)非常早期考慮規(guī)劃。早期規(guī)劃便于在階段 1 收集恰當數(shù)據(jù),加強階段 2 商業(yè)工藝確認的有效性和成功的目標,它也為階段 3 持續(xù)工藝確證建立一個基礎。
工藝驗證生命周期階段 1 之前(以及可能在過程中)可利用知識來源包括:
類似工藝(例如平臺工藝)的以往經(jīng)驗;
產(chǎn)品和工藝理解(從臨床和臨床前活動);
分析的特征描述;
已發(fā)行的文獻;
工程研究/批;
臨床生產(chǎn);
工藝研發(fā)和特征研究;
下面章節(jié)概述了工藝驗證一般生命周期方法中階段 1 的輸出,描述在圖 3.0-1 中。

3.1 第一階段工藝驗證應達到的目標
下面的列表概括了在工藝驗證生命周期中從第一階段(工藝設計)到第二階段(性能確認)轉(zhuǎn)移所需的信息。本節(jié)詳細討論應轉(zhuǎn)移的資料并提供額外的參考信息。
目標產(chǎn)品質(zhì)量概況(QTPP)-第一階段開始就完成的。
關鍵質(zhì)量屬性及對應的關鍵性風險評估和理想的置信程度
• 生產(chǎn)工藝設計
• 工藝描述:包括每個操作單元的工藝輸入、輸出、收率、中間檢測與控制和工藝參數(shù)(設定控制點和范圍);
• 工藝溶液處方、原材料及規(guī)格;
• 批記錄和來自實驗室或中試生產(chǎn)規(guī)模的生產(chǎn)數(shù)據(jù)
• 分析方法(包括產(chǎn)品、中間產(chǎn)品和原材料)
• 質(zhì)量風險評估
• 工藝特性化前對參數(shù)進行基于風險的初步分類
• 關鍵性和風險評估
• 基于關鍵性和風險分析識別工藝參數(shù)
• 工藝特性化
• 工藝特性化計劃和方案
• 研究數(shù)據(jù)報告;
• 工藝控制策略
• 放行標準;
• 中間產(chǎn)品控制與限度;
• 工藝參數(shù)設定點和范圍;
• 日常監(jiān)控要求(包括中間品取樣和測試);
• 中間產(chǎn)品、加工溶液的貯藏和工藝步驟的時間限度;
• 原材料/成分規(guī)格;
• 設計空間(如適用)
• 過程分析技術應用和算法(如果使用 PAT)
• 產(chǎn)品特性化試驗計劃(即不包括產(chǎn)品放行檢驗中的試驗)
• 生產(chǎn)技術-按工藝要求評價生產(chǎn)設備能力和適應性(也可以在第二階段 2a 中進行)放大/縮小方法(實驗室模型評價/確認)
• 藥品開發(fā)文件
工藝設計報告
• 工藝驗證主計劃
3.2 目標產(chǎn)品質(zhì)量概況(QTPP)
藥物開發(fā)的目的是設計一種在制造過程中始終達到藥品預期性能的高質(zhì)量的產(chǎn)品。藥物開發(fā)始于確定預先定義的目標。在目標產(chǎn)品質(zhì)量概況(QTPP)中應有描述。QTPP 始于第一階段開始并在整個產(chǎn)品生命周期中被參考引用。
QTPP 記錄了全部藥品相關的質(zhì)量要求。而且,還要定期更新在藥品開發(fā)過程中產(chǎn)生的新的數(shù)據(jù)。
但是,QTPP 不應偏離藥品目標產(chǎn)品概況(TPP)所建立的核心目標。
注:作為工具的目標產(chǎn)品概況(TPP)有助于申請人-監(jiān)管者的相互影響與交流。因此,TPP 包括諸如
藥物適應癥與用處、劑量與用法、劑型與規(guī)格、禁忌、警告與注意事項、不良反應、藥物相互作用、
濫用和依賴性、和過量等不在本文范圍內(nèi)的信息。
目標產(chǎn)品概況包含的相關特性有:
臨床預定用處(例如:劑型與規(guī)格、服用方法、傳遞系統(tǒng)、容器與密閉系統(tǒng))。
原料藥質(zhì)量屬性:適用于開發(fā)的藥品劑型(如物理的、化學的、和生物學的性質(zhì))。
藥品質(zhì)量屬性:適用于預期上市產(chǎn)品(例如:純度/雜質(zhì)、穩(wěn)定性、無菌性、物理和化學性質(zhì))。
•治療部分的釋放或傳遞,以及適用于藥品的影響藥代動力學特性的屬性(例如溶解、空氣動力學行為)。
輔料和成分質(zhì)量屬性、藥物-輔料相容性、和藥物-容器相容性:影響藥品的工藝能力、穩(wěn)定性或生物學作用
QTPP 總結(jié)了藥品的質(zhì)量屬性,保證藥品的安全性和有效性。 QTPP 為產(chǎn)品質(zhì)量屬性關鍵性評估提供了一個起始點。
3.3 關鍵質(zhì)量屬性
關鍵質(zhì)量屬性(CQA)是指保證預期藥品質(zhì)量的物理、化學、生物學或微生物性質(zhì)或特性在適宜的限度、范圍或分布范圍內(nèi)。CQAs 可以與原料藥、成品、輔料、中間產(chǎn)品(中間原材料)和容器/密閉系統(tǒng)有關。在工藝開發(fā)的早期可以限定對產(chǎn)品屬性有效的信息。為此,起初設定的關鍵質(zhì)量屬性可能來自早期開發(fā)和/或類似產(chǎn)品而不是大量的產(chǎn)品性質(zhì)。
質(zhì)量屬性的關鍵程度來源于利用基于風險的工具和質(zhì)量屬性對安全性與有效性的潛在影響。在對科學證據(jù)和風險進行了綜合評估之后,根據(jù)關鍵性程度對質(zhì)量屬性排序,這樣可能更能反映結(jié)構-功能關系的復雜性和屬性分類不確定性的變化程度。與 CQAs 無關的屬性在工藝開發(fā)中也應加以考慮。
CQAs 與標準規(guī)格是不同義的。另外,也沒有必要將 CQAs 與標準規(guī)格一一對應。標準規(guī)格是檢查試驗的一個列表,是分析過程的依據(jù),是待檢品可接受標準的數(shù)字限度、范圍或其他標準。被確定為 CQAs 的幾個產(chǎn)品屬性可以用單一方法檢查,因此,可以建立單一的測試標準(例如:API 的溶解度、硬度、孔隙度是可以用一個試驗-溶出度來評價的 CQAs)。有些在工藝過程中容易控制和達到的 CQAs 可以不包含在標準規(guī)格中(如病毒清除不是每批檢查),而一些不是關鍵性的屬性也可能制定在標準規(guī)格中。
潛在 CQAs 的確定是一個始于產(chǎn)品開發(fā)早期的持續(xù)性活動。它需要利用產(chǎn)品及其應用以及臨床和非臨床數(shù)據(jù)等一般知識。在產(chǎn)品開發(fā)的初期,CQAs 是易變的,所以需要質(zhì)量風險管理方法以發(fā)展產(chǎn)生產(chǎn)品和工藝的知識(有關討論見 6.1 節(jié)“風險管理的應用”)。商業(yè)產(chǎn)品的關鍵質(zhì)量屬性應在第二階段活動開始前被定義。
3.4 定義制造過程
制造過程應設計成可以持續(xù)提供滿足所需質(zhì)量屬性的產(chǎn)品。因為制造過程是在開發(fā)過程中定義的,
所以,過程描述就是用于幫助風險評估的實施和控制策略開發(fā)的工具。制造過程由一系列單元操作組成,工藝描述、方塊圖、工藝流程圖描述每個單元操作。制造過程中的每個單元操作應用相似的詳細程度進行描述。每個工藝描述應包含如下信息:
• 工藝需求,包括原材料、規(guī)模和操作指令;
• 工藝參數(shù)設定點和范圍;
• 鑒定和定量所有材料流(附加物、廢料、產(chǎn)品線);
• 測試、取樣和中間控制
• 產(chǎn)品和附加溶液的保持時間和保持條件;
• 估計分步產(chǎn)量和持續(xù)時間;
• 儀器選型,包括色譜柱和過濾單元;
•制造商(如過濾器)和產(chǎn)品組分(如玻璃瓶、瓶塞)的特殊鑒定(如制造商、零件號碼);
• 成功再現(xiàn)工藝所必須的其他信息。
圖 3.3-1 描述了一個單元操作的工藝圖實例,并在表 3.4-1 提供了一個工藝描述簡表。工藝知識及理解的發(fā)展反映在臨床批記錄中。這些都是在工藝描述中定義制作過程的重要信息源。從臨床試驗材料制造收集的數(shù)據(jù)可能有助于確定過程能力、設定標準、設計 PPQ 方案和可接受標準、評價實驗室模型和轉(zhuǎn)移工藝。知識管理的策略和原則將在第 6.5 節(jié)“知識管理”中進一步討論。
工藝描述是以報告的形式歸檔并可能編入產(chǎn)品技術轉(zhuǎn)移(TT)文件包。由于材料要求的增加(如工藝和分析開發(fā)、臨床需要),第一階段工藝可能會改變,增進產(chǎn)品了解導致 CQAs 改變,工藝的進一步了解導致單元操作的增加、刪除或調(diào)整。這些變化和支持性的說明應文件記錄下來。這些信息應保存在知識管理系統(tǒng)中。
開發(fā)報告中商業(yè)制造過程的開發(fā)和歸檔應先于正式的工藝特性研究。在工藝特性化過程中獲得的知識增加可能會要求增加工藝描述的變更。工藝的所有變更應按質(zhì)量系統(tǒng)定義的變更控制程序獲得批準。
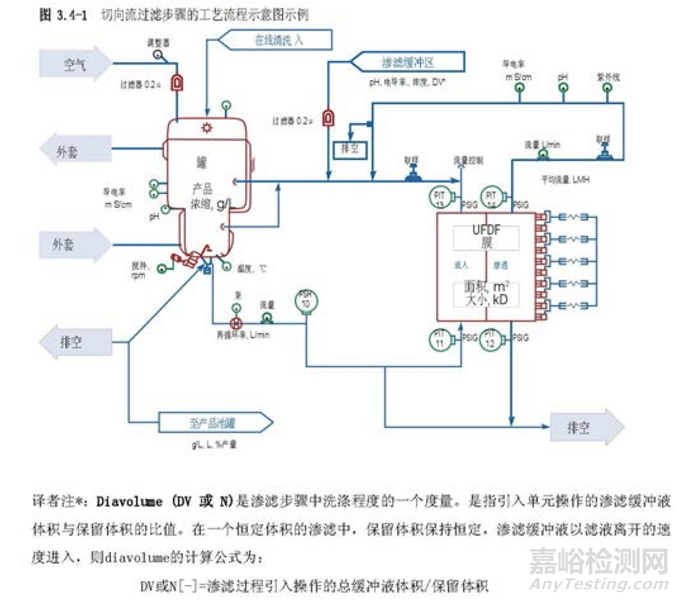
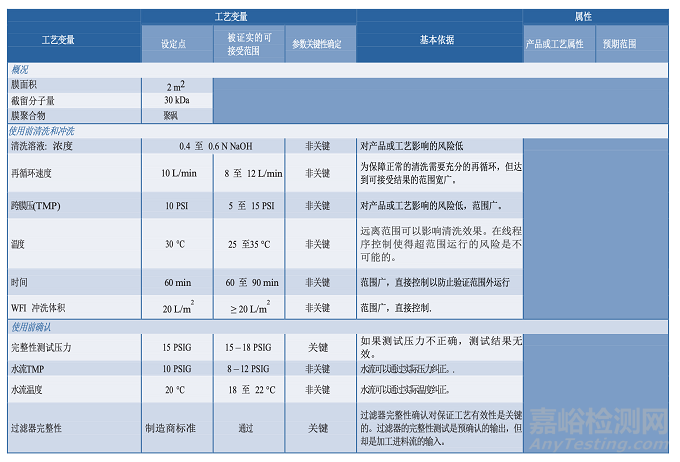
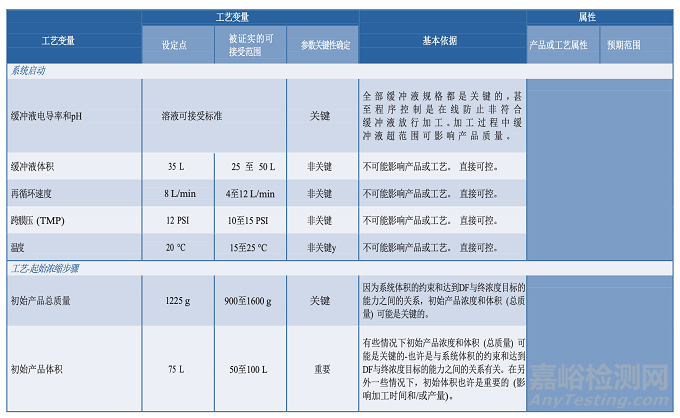
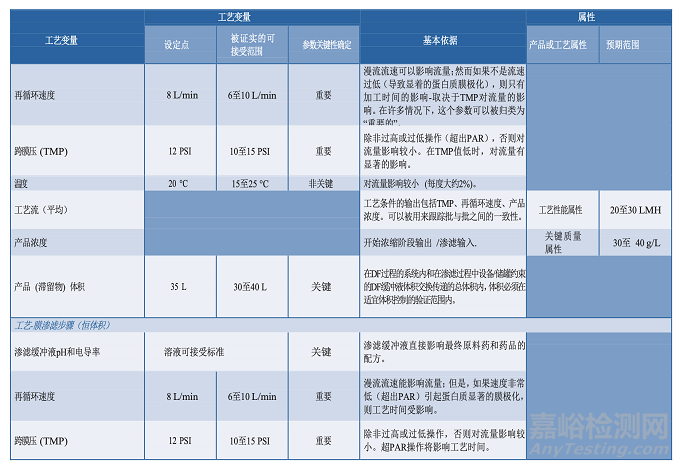
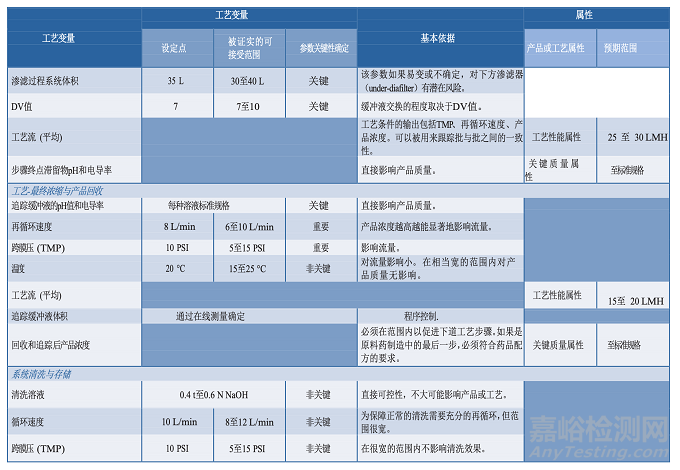
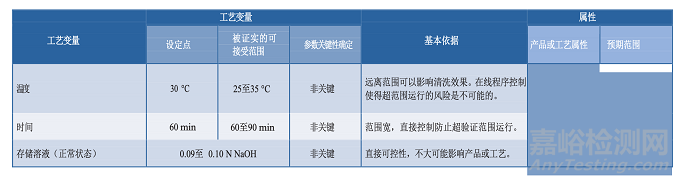
3.5分析方法
原材料、中間樣品、原料藥、藥品成品的分析是控制策略(第3.8節(jié))和工藝性質(zhì)研究的主要方面。
用于這類研究的分析方法應該適用于其預定的用途,科學合理,可靠性、重現(xiàn)性好。應頒布開發(fā)過程中使用的分析方法的確認/驗證策略,并提供在生命周期該階段測試的評價方法(27)。關于分析方法評估的指南在FDA工藝驗證指南中有論述(3)。工藝性質(zhì)研究中使用的分析方法信息應包含在工藝特性化計劃中,并記錄在研究報告中。方法確認也應該記錄在案,因為工藝性質(zhì)研究可能在開發(fā)實驗室進行,儀器必須做適當?shù)男屎途S護。
3.6 風險評估和參數(shù)關鍵性設定
風險評估在商業(yè)控制策略開發(fā)中具有重要作用。風險評估是在生命周期第一階段的幾個點上由跨學科團隊實施的,分屬于不同目標。(見第6.1節(jié)風險管理的應用)風險評估工具提供了一個結(jié)構化的方式記錄與風險評估的結(jié)果相關的數(shù)據(jù)和理由,并成為工藝開發(fā)歷史記錄的一部分。
如圖3.0-1所示,在第一階段通過質(zhì)量風險評估初步識別關鍵質(zhì)量屬性。初始質(zhì)量風險評估是識別對產(chǎn)品質(zhì)量或工藝性能影響最大的工藝輸入?yún)?shù)變量的原因和影響分類。這個評估主要是基于已有知識或早期開發(fā)工作,評估結(jié)果為下述工藝性能研究提供基礎。
了解工藝參數(shù)變化的影響和應用適當?shù)目刂剖巧虡I(yè)控制策略開發(fā)的基本要素。 ICH Q8 (R2)定義關鍵工藝參數(shù)(CPP)為:“對CQA有影響的可變參數(shù),因此,應該被監(jiān)測或控制以保證該工藝產(chǎn)生預期的質(zhì)量”(3)。
根據(jù)對工藝的影響,工藝參數(shù)可以被進一步分類。在某些情況下,工藝性能的控制和監(jiān)控是作為一個額外的控制手段,確保控制狀態(tài)的協(xié)調(diào)一致。試驗顯示對工藝性能有影響的工藝參數(shù)可以分類為重要工藝參數(shù)(KPP)。 KPPs可以影響工藝性能屬性(如在細胞培養(yǎng)過程中的抗體滴度或下游純化產(chǎn)量),但不影響產(chǎn)品關鍵質(zhì)量屬性(15)。在有些工藝中,KPPs的識別和適當控制是有用的,因為工藝性能評估可能是批內(nèi)一致性證明的一個重要手段。然而,除了普遍認可的定義ICH Q8(R2)的關鍵工藝參數(shù),工藝參數(shù)命名會不規(guī)范、方法可能會有所不同。出于這個原因,在組織內(nèi)參數(shù)命名的定義必須清楚地記錄并理解。在整個工藝驗證生命周期中參數(shù)命名的定義應保持一致。
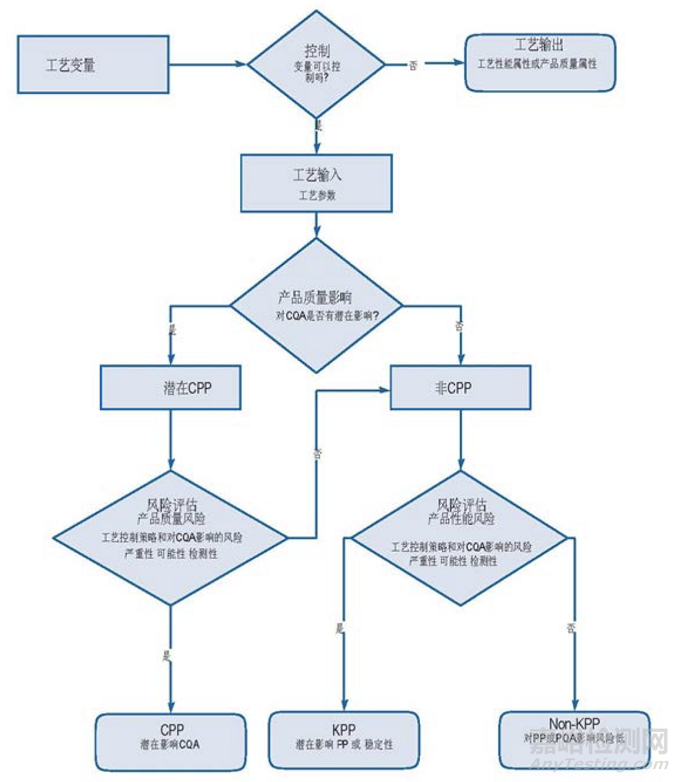
圖3.6-1提供了一個決策樹示例,以指導結(jié)合質(zhì)量風險評估進行參數(shù)命名。決策樹有助于將工藝參數(shù)分類為關鍵、重要或非重要(見定義)。決策制定工具可以幫助參與者形成共識,并有利于提高決策過程的一致性,以及作為風險評估過程部分有理由一致的記錄文檔。
決策樹可以用于來源于工藝性能研究的支持數(shù)據(jù)前后的風險評估。
• 參數(shù)或?qū)傩裕汗に囎兞靠梢允菃卧僮鞯妮敵龊蛯α硪粏卧妮斎搿σ粋€指定的單元操作,根據(jù)每個變量的直接可控性初步設定為參數(shù)或?qū)傩浴?/span>
是—直接可控的工藝輸入?yún)?shù)理論上有助于工藝的可變性。
否—不能直接控制的工藝輸出是被監(jiān)測的屬性,可能表示工藝性能或產(chǎn)品質(zhì)量。
工藝參數(shù):對關鍵質(zhì)量屬性的潛在影響。
是—如果懷疑參數(shù)的變化對CQA有影響,或如果數(shù)據(jù)顯示可能會有影響,則指定這個參數(shù)為關鍵工藝參數(shù)(CPP)。
否—參數(shù)為非關鍵工藝參數(shù)并進一步評價。
• 非關鍵工藝參數(shù):如果超定義范圍運行,潛在影響工藝性能或穩(wěn)定性。
是—參數(shù)指定為重要工藝參數(shù)(KPP)。
否—在較寬的范圍參數(shù)對工藝無影響。參數(shù)被指定為非重要工藝參數(shù)(non-KPP)。
Figure 3.6-1 關鍵性命名決策樹
3.7 工藝特征描述
工藝特性描述是一套文檔證明的研究,在該研究中操作參數(shù)被故意改變以確定它們對產(chǎn)品質(zhì)量屬性和工藝性能的影響。該方法使用來自于風險評估的知識和信息以確定一套工藝特性研究以檢驗提議的范圍和對工藝參數(shù)的交互,結(jié)果/產(chǎn)生的信息被用于確定PPQ(工藝性能確認)范圍和接受標準,它也能用于設置最終參數(shù)范圍以及能被用于發(fā)展一個設計空間(如果用一個加強方法an enhanced approach),例如包括先進的分析的和/或生產(chǎn)控制技術到工藝開發(fā)中。能設計實驗以檢查擬議的范圍以及探索一比將用于正常運行更寬的范圍。
工藝特性的一個要素可能包括多變量設計實驗以明確工藝設計空間,而單變量的方法對一些變化去建立一個證實的接受范圍是合適的,多變量研究對工藝參數(shù)/物料屬性間的交互作出了說明/解釋。由于旨在特征化工藝和為工藝參數(shù)設置可接受范圍的研究通常在實驗室規(guī)模執(zhí)行,因而實驗室規(guī)模研究預測工藝性能的能力是合適令人滿意的,當一個實驗室規(guī)模的模型被用在研發(fā)中時,該模型的充分性應該被證實和合理說明。當在實際和期望的性能間有差異時,實驗室模型和模型預測法應該適當糾正,因為從研究中得到的結(jié)論被直接應用到商業(yè)規(guī)模工藝。實驗室模型的確認是必不可少的,縮小比例的模型的確認應該證實它們的性能代表實際的生產(chǎn)規(guī)模工藝,這通過比較運行參數(shù)和輸入輸出,包括產(chǎn)品質(zhì)量屬性。對用于蛋白產(chǎn)品的色譜層析步驟的縮小比例的模型能通過在設置點輸入?yún)?shù)執(zhí)行多次運行以及比較與實際規(guī)模的單元操作間的結(jié)果來確認。
評價的參數(shù)應該包括那些影響工藝一致性,諸如工序收率、洗脫圖、洗脫體積和/或保留時間,然后這些應該與那些代表產(chǎn)品質(zhì)量諸如可憐的純度和工藝有關的水平以及與宿主細胞有關的雜質(zhì)結(jié)合起來。代表商業(yè)生產(chǎn)工藝的小分子中試規(guī)模模型可能被用于支持PPQ數(shù)據(jù),在固體和液體口服劑型,商業(yè)批量的10%和/或100000單位曾被考慮為一個代表性規(guī)模。某個工藝的放大的效果,諸如混合易溶解物質(zhì)、藥片壓縮、或液體灌裝可能眾所周知。在原液10%的批量或運行100000劑量單位的倍數(shù)提供一個充分持續(xù)時間以確定控制程度和工藝特征,而未覆蓋任何初步的主要問題。當小規(guī)模研究被用于支持PPQ時,實際規(guī)模確認/評估可能被執(zhí)行。對于縮小比例研究,其原料、組件屬性、設備以及工藝參數(shù)應該是具可比性和能表明預期用于商業(yè)產(chǎn)品的工藝。
3.8 產(chǎn)品特征測試計劃
一些產(chǎn)品特性可能作為日常放行測試標準的一部分而不必測試,此類產(chǎn)品特性例子包括生物技術產(chǎn)品的殘留DNA水平(當DNA清除率已經(jīng)建立在一個能清晰超出安全要求的水平時)或固體口服制劑的最終產(chǎn)品的多孔性(當執(zhí)行溶解度測試時),除了放行標準,階段1可交付的成果應該包括其它對DS、DP或關鍵的中間體的測試為了說明對產(chǎn)品和工藝的全面理解。
3.9 控制策略
建立一個有效和適當?shù)墓に嚳刂撇呗允请A段1中藥物研發(fā)最重要產(chǎn)出之一。一個恰當?shù)目刂撇呗允腔谠陔A段1中獲得的知識和經(jīng)驗,它的效力關系到生產(chǎn)工藝保持受控狀態(tài)的程度。正如上面討論的階段1的另一個方面,一個有效的工藝控制策略的開發(fā)時一個迭代過程(自我循環(huán)過程),它開始在研發(fā)早起以及成為工藝和產(chǎn)品知識增加。一個具耐受性的控制策略包含工藝中單個單元操作的所有元素,所有產(chǎn)品質(zhì)量屬性和工藝參數(shù),不管它們是否被歸類為關鍵,都將包括在一個完整的工藝控制策略中,完整的工藝控制策略包括下列元素:
原料控制
確保始終如一的輸出的管理輸入(原料和組件)質(zhì)量的能力是工藝控制策略必不可少的,輸入應該基于它們引入變化或污染到產(chǎn)品和/或工藝中的潛在風險而進行分類。產(chǎn)品變化性可能包括CQAs的改變,反之工藝變化性可能包括收率不一致、反應動力學、過濾性或其它非產(chǎn)品質(zhì)量相關影響。對于生產(chǎn)工藝中所用的許多原料,選擇適當級別(基于純度、化學和物理特性、和/或微生物標準例如內(nèi)毒素)可能是一個充足的控制水平;對于高風險原料,理解其對產(chǎn)品和工藝變化性的促成程度對建立那些物料的標準是必不可少的,一旦這關系被理解,適當?shù)娘L險壓縮步驟能采用成為控制策略的部分(見6.1.4節(jié))。
過程和放行標準
過程和產(chǎn)品標準可能與產(chǎn)品安全性和有效性相關或可能保證產(chǎn)品一致性,確認不符合產(chǎn)品標準(過程或產(chǎn)品)取消臨床或商業(yè)使用物料資格。關于設置標準的指南在ICH指南文件Q6a和Q6b中提供。
過程控制
過程控制是工藝的輸入以及在在生產(chǎn)過程中執(zhí)行檢查以監(jiān)控和調(diào)整工藝(如果適當),和/或確保中間體或產(chǎn)品符合標準或其它規(guī)定質(zhì)量標準。
性能參數(shù)
性能參數(shù)(例如:片劑/膠囊崩解、 收獲期或生長高峰期的細胞密度/活力)是工藝輸出,不能直接控制單它反映工藝是否按預期。
工藝參數(shù)設置點和設置范圍
工藝參數(shù)可變性對每個單元操作的輸出和最終產(chǎn)品的影響的知識在工藝發(fā)展和工藝特征化過程中演變(3.7節(jié)),這信息連同工藝設備能力(4.1節(jié))被用于建立參數(shù)設置點和范圍(包括警戒限和行動限范圍),也可用于評估由參數(shù)漂移所致的工藝偏差的嚴重性。參數(shù)范圍可能被指定為正常操作范圍(NORs),或當有支持數(shù)據(jù)證實時作為已證實的可接受范圍(PARs)。
工藝監(jiān)控(數(shù)據(jù)審核、取樣、測試)
工藝監(jiān)控包括測量數(shù)據(jù)(例如流速、溫度、體積、pH)、過程取樣計劃、以及適當分析檢測。數(shù)據(jù)收集和分析在階段1開始以及是階段2不可缺少的部分。數(shù)據(jù)收集工作最終發(fā)展成為階段3中描述的持續(xù)工藝監(jiān)控程序(見5.0節(jié))
加工和保留時間
對所有工藝中間體(或過程物料)、藥物成分、原液藥物產(chǎn)品以及準備的溶液的保留條件和時間是工藝控制策略一個不可或缺的部分,應執(zhí)行研究以支持這些限度,對工藝步驟的時間限度也應該是控制策略的一部分。
過程分析技術(PAT)
過程分析技術(PAT)是執(zhí)行控制策略的一個方法,使用PAT,CQAs被實時監(jiān)控(用在線分析),在生產(chǎn)過程中其結(jié)果用于調(diào)節(jié)CPPs以減少產(chǎn)品變化(CQAs)或得到在期望范圍內(nèi)的變化性小的已知的CQAs。
PAT使用產(chǎn)品和工藝知識以及設備自動化和分析儀表技術,成功的PAT應用要求一個徹底的特征化工藝(3.7節(jié)),在該特征化工藝中,CPPs和CQAs間關系用數(shù)學模型探索,諸如多變量分析。對控制策略的理解的應用也影響生產(chǎn)工藝中測控系統(tǒng)的確認。為支持PAT的執(zhí)行,階段1交付必須描述CQA監(jiān)控計劃和基于工藝響應調(diào)節(jié)CPPs的算法。設備、 測量系統(tǒng)以及工藝的確認(階段2)必須證明安裝建立的運算法則調(diào)整CPPs的能力以及確認這些調(diào)整導致的可接受的和預測的輸出。因而,基于控制方法的PAT需要被確認。
3.10臨床生產(chǎn)經(jīng)驗-批記錄和生產(chǎn)數(shù)據(jù)
在階段1過程中,臨床批被用于臨床試驗中以支持產(chǎn)品批準,這數(shù)據(jù)可能連同正式的工藝特征數(shù)據(jù)一起被用于支持生產(chǎn)工藝參數(shù)和工藝控制策略的建立,這些數(shù)據(jù)也包含PPQ之后將繼續(xù)的工藝監(jiān)控的起始數(shù)據(jù)。早期的批數(shù)據(jù)可能不包括所有在最終商業(yè)工藝中執(zhí)行的控制,但這信息對評估工藝性能仍然有價值。如果用于支持范圍和限度,臨床批數(shù)據(jù)應該包括在最終工藝設計報告中用于證明工藝和控制策略。最終批記錄應該在階段1結(jié)束時產(chǎn)生,它們將支持最終的商業(yè)工藝和充當階段2的前奏。
在一些情況下,用來自于臨床批的數(shù)據(jù)支持PPQ階段2可能是適當?shù)模@個方法的原理應該被記錄且包括在工藝驗證主計劃中。
3.11工藝設計報告
工藝設計報告也是階段1的輸出,作為一份詳細描述預期的商業(yè)工藝的動態(tài)文件,它可能在內(nèi)部程序里有各種各樣的主題名稱。階段1研究數(shù)據(jù)被用于支持這個文件以及證明范圍和工藝控制策略。生產(chǎn)工藝變更時收集到的額外的數(shù)據(jù)和工藝知識收集起來病囊括在階段2和3中工藝設計報告應該被更新以包括這信息的更新,這全面的文件包括:
• 參考CQAs和支持風險評估
• 工藝流程圖
• 工藝描述表
• 輸入(過程控制)
• 輸出(過程測試和限度,過程標準)
• 工藝參數(shù)和范圍;
• 對CQAs和工藝性能有影響風險的參數(shù)分類
• 設計空間,視情況而定;
• 支持所有參數(shù)范圍的合理說明和數(shù)據(jù)(例如,特征數(shù)據(jù)、開發(fā)研究、臨床生產(chǎn)歷史);
3.12工藝驗證主計劃
一個工藝驗證主計劃可能在階段1到準備階段2活動期間被發(fā)起,它應該描繪概述驗證策略和支持原理,通常包括:
• 工藝特征計劃;
• 生產(chǎn)工藝和控制策略的描述;
• 部門與責任;
• PQ和PPQ計劃
PPQ策略(例如單個單元操作或單元操作的合并、bracketing、家族法或矩陣法)和單個草案、可適
用的輔助研究清單(例如混合培養(yǎng)基準備、過程持續(xù)時間、樹脂生命周期);
所用的設備和設施清單;
• 分析方法和它們狀態(tài)清單;
• 取樣方法;
• 在計劃下將被執(zhí)行的草案列表;
• 建議的時間表和交付計劃;
•處理偏差和再版的程序;
• 持續(xù)工藝確證計劃;
3.13 階段 生產(chǎn)和技術考慮
生產(chǎn)設備的能力和程序?qū)S護工藝參數(shù)在預設定限度里的能力有關鍵影響,工藝設備的測量和控制是階段2科目之一,工藝確認,4.1部分有介紹。設備確認活動應該確認設備對預期用途的適宜性。
工藝物料流與設備和他們接觸的材料(例如聚合膜、橡膠、免洗袋和其它塑料部分)的相容性要能保證產(chǎn)品的安全和有效。產(chǎn)品接觸材料和溶出物、萃取物需要評價他們的相容性。這個工作在第一階段開始,可能包括一些需要長的前置時間的研究,并且應該結(jié)合階段二來完成。
工藝物料流與設備表面的相容性是對在生產(chǎn)接觸時他們的反應、吸收和穩(wěn)定性的衡量。相容性測試是為了證明設備表面的材料性質(zhì)不會因接觸溶液或其它產(chǎn)品相關物料而改變。另外,接觸材料也不應該改變工藝溶液或物料(通過吸附產(chǎn)品組分或者過量吸出而摻混產(chǎn)品)萃取物是一種材料的成分(例如用于藥品生產(chǎn)或儲存的產(chǎn)品接觸表面),通過使用一種夸大的外力(溶劑、時間、溫度)而得到。浸出物是來自工藝設備或存儲容器的接觸材料的成分,它們在正常使用狀態(tài)下轉(zhuǎn)移到產(chǎn)品中。
來自于用于藥品生產(chǎn)、儲存、包裝得聚合膜盒的組件(塑料存儲器、濾器、內(nèi)包裝材料、墊片、O形圈)的浸出物的種類和數(shù)量必須通過文件列明以此保證產(chǎn)品不會被摻入雜質(zhì)。要結(jié)合文獻查閱、
風險評估和實驗室研究來說明浸出物。各種確定測試的程度和浸出物種類的鑒別。還有可接受水平的設定的方法都已經(jīng)出版。
“口服吸入和鼻噴藥物的萃取物和浸出物的安全閾值和良好實踐”
“產(chǎn)品接觸表面的萃取物的評價”
“使用 QbD對萃取物 /浸出物評估的原則:為儲存在塑料包裝系統(tǒng)的最終滅菌的液體藥物確立設計空間”
“原料藥的浸出物的評價”

來源:文亮頻道


