您當前的位置:檢測資訊 > 法規標準
嘉峪檢測網 2022-07-11 23:15
[摘要] 本文梳理了美國FDA非處方藥專論制度的發展背景、歷程、主要內容和改革現狀,對美國非處方藥上市程序、非處方藥專論制定及監管法規進行分析與解讀,為我國非處方藥分類注冊管理工作提供參考。
非處方藥( over-the-counter,OTC) 是指不需要憑醫生處方,消費者即可自行購買和使用的藥品,具有治療目標明確、療效確切、安全性好、使用方便等特點。我國自 2000 年開始實施處方藥、非處方藥分類管理,主要對藥品上市后的環節進行管理[1-2]。對于上市前審批環節,我國OTC藥物的審批程序和技術要求體系與處方藥基本相同,OTC上市申請有2種方式: ① 在藥物注冊申請的同時附加非處方藥申請。② 由已上市的處方藥經評價后轉換為非處方藥的方式。另外非處方藥均需要核準說明書,比處方藥的審批流程更加嚴格。美國實行藥品分類管理的時間較早,制度體系也相對完善,在OTC上市注冊和管理上積累了豐富的經驗。非處方藥在美國除了常規的藥物上市申請程序,還可以通過簡化注冊為非處方藥的專論程序。美國OTC專制度自1972 年開始實施,其發展歷程較具有代表性。本文通過對美國OTC專論制度的建立和發展及OTC專論的主要內容、實施和改革情況進行梳理分析,為我國OTC分類注冊管理工作提供參考。
1、 美國OTC專論制度的建立和發展
OTC在美國市場上市有2種監管途徑,分別為藥物申請程序( drug application process) 和非處方藥專論程序(OTC drug monograph process)[3]。1972年美國FDA建立OTC藥物審評制度,以評估當時市場上數十萬 OTC 藥品的安全性和有效性[4]。由于OTC藥品種類繁多,美國 FDA 采用按治療類別分組的方式進行分類審評,發布OTC專論( OTC專著,OTC monograph) 。OTC專論是每個治療類別的“規則手冊”( rule book) ,內容包含活性成分、劑型、規格、說明書及對某些治療類別的包裝或檢測要求[5]。根據該手冊,OTC藥物通常確認是安全有效的,若符合既定適應證下OTC專論的要求和其他相應法規,無需新藥申請流程和美國FDA上市前批準,備案后即可銷售。根據 OTC專論程序上市銷售的非處方藥產品稱為OTC專論藥物。目前美國OTC專論包含約800種有效成分,涉及1400多種不同用途,授權超過100 000種藥物。
2020年3月之前,OTC專論的增加、刪除或更改只能通過繁瑣的三階段公共規則制定程序( notice-and-comment rulemaking) ,導致美國 FDA 在處理重大安全問題時無法快速對OTC專論進行修訂。2020年3月27日,美國總統簽署了748 號法案( H. R. 748) 《冠狀病毒援助、救濟和經濟安全法》( the Coronavirus Aid,Relief,and Economic SecurityAct,CARES) ,其中包括 OTC 專論改革的法律規定,旨在提高OTC藥品審評流程的效率、及時性和可預見性,被認為是OTC專論藥物開發和審評過程的現代化改革[6]。
2、 美國 OTC 專論制定的程序
納入OTC專論的藥品遴選審評依照美國聯邦法規第21篇“食品與藥品”條款( Code of FederalRegulations,Title 21 Food and Drugs,21CFR) 第 330部分“通常公認為安全有效的非處方藥( over-the-counter human drugs which are generally recognized assafe and effective and not misbranded) ”進行,包含兩部分[7]。其中,A 部分為一般條款( subpart a-generalprovisions) ,內容有4條,分別是330. 1 遴選安全有效非處方藥的基本條件、330. 2 妊娠期使用警告、330. 3 固體口服劑型藥物產品的識別碼標記、330. 5藥物類別; B 部分為行政管理程序( subpart B: admin-istrative procedures) ,內容有 6 條,分別是 330. 10 OTC藥物遴選分類和 OTC 專論建立的程序、330. 11 新藥上市申請( NDA) 偏離適用的專論、330. 12 在之前藥物療效研究( DESI) 中已審查的 OTC 狀態、330. 13根據 OTC 審評建議以 OTC 上市使用的條件、330. 14將藥物分類為公認安全有效 OTC 藥物的標準和程序、330. 15 FDA 審評工作時限表、申請范圍和安全性有效性數據提交。
OTC 專論建立的程序在 21CFR 330. 10 條款中包含 12 個程序,分別為組建審評專家委員會[8]、數據遞交和審查、審評專家委員會審議、安全有效性和說明書標準、審評專家委員會提交報告、擬納入專論初稿、審評專家委員會提交完整報告、專論意見稿、專論正式稿、行政記錄、法院上訴、專論修訂。OTC 專論審評共有 3 個階段狀態。第 1 階段為擬納入專論初稿( advanced notice of proposed rulemaking,ANPR) ,由審評專家委員會審議后認可其安全性和有效性,并出具審評聲明和推薦的說明書,形成報告提交給局長,并征求意見。審評專家委員會一般將遴選藥品分成 3 類: Ⅰ類為對聲明的適應證用途公認是安全有效的 ( generally recognized as safe and effective,GRASE) ,被稱為GRASE藥品,經進一步審議后可納入 OTC 專論。Ⅱ類為對于聲明的適應證用途來說不能被公認為安全有效的( not generally recog-nized as safe and effective,NGRASE) ,不能作為 OTC專論產品進行管理。Ⅲ類為安全有效性證據不足的,暫時不能進行分類[9]。第 2 階段為專論意見稿( tentative final monograph,TFM) ,美國 FDA 基于專家委員會對成分的審查和公眾意見及數據資料等對藥物活性成分的審查,形成TFM 發布擬認定為安全有效且不含商標的OTC產品,并進行為期60d的公示。第3 階段為專論正式稿( final monograph,FM) ,OTC 專論正式稿中OTC成分及適應證是被認定安全有效的,最終在美國聯邦法規( CFR) 中建立條款。
目前的OTC專論正式稿收錄在21CFR第 331 ~358 部分,為OTC專論藥物提供合法上市的資格。在21CFR 310. 545 中列出了NGRASE 活性成分,即非OTC專論的活性成分。21CFR 310. 519 ~ 548 中所列的成分是需要新藥審批的,也不是OTC專論的活性成分。部分OTC產品的TFM被修改多次,增加了新適應證或用途,刪除了一些轉移到其他專論中的成分,或者納入了新的數據和信息而引發變化,導致整個專論最終沒有完成出版。OTC專論可根據需要通過提交申請進行修訂。在TFM公示期結束后,OTC專論正式稿發布前,若提交新的數據和信息將被視為對專論的修訂,并且需在OTC專論正式稿發布后才會被考慮。
在美國 FDA 網站上,列出了OTC產品和活性成分參考,提供了按活性成分字母排序、按專論 /類別字母順序和總表格3個版本,以方便快速查詢[10]。
3、美國 OTC 的上市申請
美國OTC上市注冊申請有OTC專論程序( OTCdrug monograph process,21CFR 330 ) 和NDA程序( 21CFR 314)[9],既可以遞交OTC專論申請,也可以遞交 505( b) ( 1) ,505 ( b) ( 2) ,( 505) ( j) 申請。無論哪一種申請程序,OTC 藥品必須符合安全性和有效性明確的標準。盡管2 種申請程序會有不同的要求,但2種申請程序和其他申請程序相比,并沒有要求更高的安全有效性標準。在這2種申請程序下,產品都必須按照21CFR 210中定義的現行良好生產規范( current Good Manufacturing Practices,cGMP)進行生產,并且必須符合 21CFR 201. Subpart C中的說明書內容和格式要求[11]。
OTC 專論程序是基于專論中包含的GRASE 活性成分,與基于藥物處方的 NDA 申請不同。在活性成分、適應證、說明書等方面均符合OTC專論申請的非處方藥,且符合cGMP 要求,不需要經美國FDA特別批準就可以上市。例如,如果OTC防曬藥物產品包含符合OTC防曬專論中認可的配方、說明書和檢測的標準,即可以合法上市銷售。有偏離或不符合OTC專論要求的非處方藥,在上市前需要審批,可通過NDA申請( 21CFR 330. 11) 或公民請愿( citizen petition,21 CFR10. 30) 途徑進行。提交公民請愿申請不需要支 付《處方藥使用者費用法》( Prescription Drug User Fee Act,PDUFA) 規定的費用,內容是公開的。通過NDA申請需要支付PDUFA規定的費用,其內容是保密的,且根據PDUFA規定,有進度時限要求,批準后可獲得市場專營期。
對于在OTC專論制度施行前已在美國上市的,或尚未在美國上市的OTC藥品,在 21CFR 330. 14“OTC藥物分類的其他標準和程序”中規定,如果要考慮納入 OTC 專論進行管理,必須滿足 2 個條件:① 在市場上以OTC藥品來銷售。如果是在其他國家或地區銷售某一類OTC藥,必須僅在藥店出售,有明確證據表明其活性成分、用法用量等所呈現的毒性或其他潛在危害都是有限的。② 在同一國家或地區至少連續銷售5年,且數量達到一定規模。如果一個國家連續5年上市后的數據不充分,可能需要補充1個或更多國家上市后的數據。
如果將現有的NDA 處方藥整體轉變為OTC,不變化劑型和給藥途徑,可以遞交有效性的補充申請。如果是將現有的處方藥部分適應證轉變為OTC,需要遞交 505( b) ( 1) 申請。如果擬上市新的OTC產品,其活性成分、適應證、劑型之前都沒有以OTC上市,也需要通過遞交 505( b) ( 1) 或 505( b) ( 2) 申請。一般情況下,具有原研地位的參照藥物( referencelisted drug,RLD) 還在市場上銷售且已轉為 OTC 的,相應仿制藥( abbreviated new drug application,ANDA)或改良藥轉為OTC; RLD 是處方藥的,相應 ANDA還是處方藥。一般不允許同一種活性成分同時在市場上存在處方藥和 OTC 狀態,除非兩者之間存在有意義的差異,處方藥只有在執業醫師的監督下使用才安全。
對于含有2個或2個以上成分的非處方藥,在21CFR 330. 10( a) ( 4) ( iv) 的組方規則中規定,每種活性成分必須均支持適應證所聲稱的療效; 活性成分的復方組合不降低任何單個活性成分的安全有效性; 復方組合應具有組合治療的合理性。
當對產品成分和適應證進行OTC審評,但是未形成專論正式稿時,此類產品可以上市,但是按照21CFR 330. 10,此類產品上市是有風險的,擬定的GRASE條件可能會變化,美國FDA也有可能不接受專家委員會的建議,那么產品則需要進行重新標簽、召回或其他監管行動。
OTC 藥物的 NDA 申請和 OTC 專論要求不符的情況,在NDA偏離適用專論條款 21CFR 330. 11 指出,應采用 21CFR 314. 50中要求的格式,還應包含一份聲明,說明產品除了申請中提出的差異,均符合適用專論的其他所有條件。
4、美國 CARES 法案下 OTC 專論制度的改革
4. 1 OTC 專論制度的改革歷程
在美國OTC藥物一直是滿足大眾日常健康需求的有效且低成本的藥物,在醫療體系中扮演著越來越重要的角色。在美國市場上銷售的OTC藥物大多數是通過OTC專論制度上市使用的。OTC專論在一定程度上有利于該類產品順利上市使用,企業不僅可以快速上市簡單仿制類產品,還可以根據市場情況在口味、劑型等方面進行部分改良創新,從而更好地滿足了消費者多樣化的產品需求,也有利于減輕行業的監管負擔。然而,建立或修改OTC專論的法規繁瑣,規則制定的步驟復雜; 而美國FDA又缺乏足夠的經費和資源用于OTC專論的制定。目前仍有大量專論申請審評被長時間擱置,未能確定。OTC專論體系的更新缺乏及時性和靈活性,特別是在安全性方面,許多專論活性成分安全標簽變更尚未進行,科學認知的不斷發展和市場條件的不斷變化給OTC專論制度帶來挑戰。為了更好地為患者、消費者和行業服務,美國FDA對OTC專論制度進行改革,以簡化審評活動[12]。OTC專論改革的目標和需解決的問題包括: 通過用行政命令代替規則制定來改善OTC專論制定的流程; 提高審評效率、及時性和可預見性,促進創新發展; 建立可快速更新安全性內容的流程,完成待定的 OTC 專論; 提供用以支持OTC專論工作的經費支持[13]。
2015 年美國FDA開始與業界探討改革OTC專論制度并公開征求意見,于 2017年6月將最終提議遞交美國國會。2019年美國國會參議院提出的269 號《OTC專論創新改革法案》( Over-the-counter Monograph Safety,Innovation,and ReformAct of 2019) 獲得全面批準,此項法案在OTC 專論制度上進行了重大改革: ① 以行政命令方式確定OTC專論的正式稿,取代以往聯邦法規發布形式,這使得專論流程制定和發布更高效。② 建立OTC專論藥物使用者費用計劃( OTC monograph drug userfee program,OMUFA) ,該計劃從 2021 財政年開始收取費用以支持專論的審評及OTC藥物相關的檢查等工作。③ 明確OTC產品專利權,將授權對符合條件的OTC 進行為期18個月的市場專營期。
2020 年正值新型冠狀病毒肺炎疫情期間,美國國會于 3 月 27 日緊急通過CARES法案實施改革,推進OTC專論藥物現代化監管方式。CARES法案對OTC專論制定、修訂補充的行政命令程序進行了改革,還賦予美國FDA評估和收取專門用于 OTC專論藥物工作的 OMUFA 費用的權力,以幫助該機構及時開展重要的監管活動,助力更多創新性OTC專論藥物上市使用。CARES 法案實施后,新OTC專論制度體系被認為響應程度更高、更為透明,同時可持續確保相關藥品的安全性與有效性。
4.2 CARES 法案之后的OTC藥物審評
CARES法案之后OTC藥物審評不變的內容包括: OTC活性成分的審查、按治療類別分組的有效成分、GRASE的確定、符合OTC專論和其他適用要求的藥物可在未經美國FDA批準的情況下進行銷售、程序包括公開征求公眾意見。新法案在行政命令程序( administra-tive order process) 、OTC 專論命令請求( OTC mono-graph order request,OMOR) 、解決現有OTC專論藥物積壓申請( 包括以前接受過 TFM 和ANPR 的藥物、劑型微小變更的流程) 、某些OTC專論藥物市場專有期、OTC專論使用費、正式會議等方面進行了改革。
用于代替規則制定過程的行政命令流程更快速。行政命令程序可以添加、刪除或更改OTC專論的GRASE 條件,可以由行業或美國FDA發起。對于行業發起的申請( industry-initiated order) 分為第1級和第2級,可提交OMOR 以請求美國FDA啟動行政命令處理,并規定了相應的審評時間( 見表1) 。
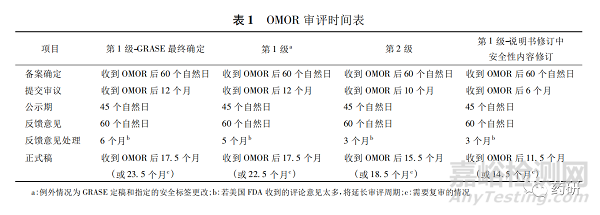
4.3 CARES 法案之后的OTC專論修訂
新法案實施后,若OTC專論存在安全性方面的問題,美國FDA可以通過啟動行政命令程序來及時修訂處理。例如,美國FDA可以更改藥品說明書標簽 ( drugfacts label,DFL) 中包含的標簽語言; 美國FDA可以發布某些具有包裝要求的行政命令,以鼓勵按照說明書標簽使用。在特定情況下,美國FDA可啟用加急行政命令( expedited administrative orders) 。
4.4 CARES 法案之后的OTC專論工作計劃
美國FDA在2021年10月1日前開始發布第一個工作計劃,隨后工作計劃的發布頻率不低于每年1次。每年美國FDA都會發布1份非約束性的OTC專論藥物問題清單,并計劃在未來3年內解決這些問題,包括安全標簽的更改。對于需要提交數據的問題,美國 FDA 將在其上注明提交日期。
美國FDA在2021財年向公眾開放OTC Mono-graphs@ FDA數據庫,公眾能夠在此搜索查找提議和最終的行政命令,增加、刪除或更改OTC藥物專論的條件; 同時公眾也可在此對擬提議的行政命令提出意見[12]。
5、對我國 OTC 分類注冊管理的啟示
對我國OTC分類注冊管理的啟示非處方藥OTC專論在越來越多的國家或地區的監管實踐中被運用,該項舉措既有利于節約審評資源、提高監管效率、加快 OTC藥物上市速度,也可以滿足公眾對于OTC藥物的需求。美國對采用專論程序上市的產品不進行傳統意義上的審評審批,看似放松了對這類產品的注冊監管,弱化了上市準入環節的風險管控,而事實并非如此,OTC專論制定過程本身就是風險管控的過程。從美國OTC專論制度的建立發展和近期的改革舉措可以看出,OTC上市的注冊管理是整個OTC藥品管理制度體系的組成部分,OTC專論制度的實施必須配合完善的藥品全生命周期風險管理制度和措施。
我國2020年頒布的《藥品注冊管理辦法》( 以下簡稱《辦法》) 首次明確了國家藥品監督管理局藥品審評中心( 以下簡稱藥品審評中心) 制定符合OTC 藥品特點的上市工作程序和技術指導原則,是對OTC藥品審評管理的一大進步,將有利于促進更多的OTC上市,同時有利于加快OTC上市速度。《辦法》第十五條中指出“處方藥和非處方藥實行分類注冊和轉換管理。藥品審評中心根據OTC的特點,制定OTC上市注冊相關技術指導原則和程序,并向社會公布。藥品評價中心制定處方藥和OTC上市后轉換相關技術要求和程序,并向社會公布”。《辦法》第三十六條指出有符合以下 4 種情形之一的,可以直接提出OTC上市許可申請: 境內已有相同活性成分、適應證( 或者功能主治) 、劑型、規格的OTC上市的藥品; 經國家藥品監督管理局確定的OTC改變劑型或者規格,但不改變適應證( 或者功能主治) 、給藥劑量以及給藥途徑的藥品; 使用國家藥品監督管理局確定的OTC的活性成分組成的新復方制劑; 其他直接申報OTC上市許可的情形。另外還規定,藥品審評中心根據藥品注冊申報資料、核查結果、檢驗結果等,對藥品的安全性、有效性和質量可控性等進行綜合審評,OTC還應當轉藥品評價中心進行OTC適宜性審查。OTC的藥品注冊證書還應當注明OTC類別。OTC適宜性審核時限為30d[14]。2020年7月藥品審評中心公開征求《化學藥品非處方藥上市注冊技術指導原則( 征求意見稿) 》意見,基于我國OTC注冊申報特點,制定OTC上市許可申請的技術要求,執行與處方藥上市許可申請一致的原則。該指導原則明確指出僅適用于化學藥品類OTC[15]。
我國的OTC產品既有化學藥品,也有中藥。目前我國OTC約5065個品種,其中化學藥品1115個品種,中藥3950個品種,中藥品種遠遠多于化學藥品。在美國銷售的OTC約有300000 種[9],且很少涉及中藥,品種數量是我國的近 60 倍。隨著我國改革的深入發展,我國已成為全球最大的醫藥消費市場之一,近年來我國OTC藥物公眾需求的不斷增加,OTC市場規模也在持續增長,但藥品審評資源非常有限,OTC注冊管理體系尚未完善。在藥物審評審批制度改革中,我國正處于仿制藥一致性評價的關鍵時期[16],按照處方藥的工作程序和技術要求,制約著OTC注冊申報和審評審批進度,OTC仿制藥的參比制劑難以確定,由于審評資源和監管資源有限,增加了審評的壓力,加重了行政審批的資源負擔。
OTC在促進公眾健康方面發揮了重要作用,且有助于節約醫療成本、減輕醫療壓力,具有較大的潛在社會價值。我國應進一步完善OTC管理的法規體系、注冊路徑和審評程序,完善配套政策,合理配置審評資源。例如,探索適合我國現階段藥品審評審批改革發展要求的OTC專論制度; 完善OTC藥物從研發、審評到上市后管理的全生命周期監管法規要求; 設置獨立的OTC政策研究和審評及上市后的監管部門; 探索建設區別于處方藥審評的OTC藥物尤其是仿制藥申報的審評程序和技術要求等,為OTC的上市使用提供更高效的途徑,以促進我國OTC的研發和使用,助力實現健康中國的戰略目標。

來源:中國新藥雜志


