您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2020-09-25 09:13
前情簡述
近年來,有一個名詞頻頻出現在人們視野——PM2.5(英文名稱:FineParticular matter),系指環境空氣中空氣動力學直徑小于或等于2.5微米的顆粒物。由于其較高的表面活性使其表面容易附積更小的有害物質與此同時較小的粒徑可以使其長期懸浮于空氣當中。自然界以及人類的一系列活動都會產生PM2.5顆粒,我國的一些區域深受其影響,給當地人的生活質量帶來嚴重的挑戰。當其在空氣中的濃度越高時所代表的空氣質量就會越差,為何會有如此論斷,系基于對人體呼吸系統的理解發現,當細顆粒的空氣動力學粒徑小于2.5微米時這類細顆粒容易伴隨著人的吸氣過程進入肺內進而沉積在肺部引發一系列病變。本文則是將上述PM2.5更換為藥物顆粒,從設計目的、體內吸收過程、產品開發重點以及國內外法規等方面來簡要闡述以肺部為主要吸收環境的藥物遞送系統。
基礎知識——干粉吸入制劑
干粉吸入劑(DryPowder Inhaler)又名吸入粉霧劑,是將一種或多種微粉化藥物與載體組成粉體混合物灌裝儲存于膠囊或泡囊當中,經特殊給藥裝置處置之后伴隨著吸氣過程產生的氣溶膠實現藥物的肺內沉積進而發揮藥物治療作用的一類新型制劑;干粉吸入劑設計初期主要應用于哮喘、慢性肺阻塞、肺部感染等肺部疾病的靶向治療,經臨床使用價值的不斷肯定該技術已經發展成為以肺部為給藥環境實現全身治療作用的藥物傳遞系統。
基礎知識——呼吸系統
DPI是基于口腔吸入的給藥劑型,只有從呼吸的生理機理上理解藥物的吸入過程才能實現對產品的深刻理解。
呼吸過程簡述:吸氣時,伴隨著的胸腔張肺內壓強減小,基于此壓差空氣被吸入肺部,對于DPI而言在此壓差下形成氣溶膠實現藥物的肺內沉積(一般情況下,該壓差大約為4kPa);呼氣時,胸腔收縮、肺內壓強增大,大于外部壓強進而使肺內氣體排出,對于空氣動力學粒徑較小的顆粒在吸氣過程中不易沉降在肺內故而伴隨著呼氣過程外出體外。
基于對呼吸過程的理解以及成像技術的支持,確定了空氣動力學粒徑與體內沉降部位的一系列關系。
|
呼吸系統 |
沉降粒度范圍 |
|
咽喉部位及其以上 |
約≥5μm |
|
氣管(主支氣管) |
約3~5μm |
|
次級支氣管 |
約2~3μm |
|
支氣管末端 |
約1~2μm |
|
肺泡 |
約0.5~1μm |
空氣動力學粒徑小于0.5μm的細顆粒一般會伴隨著呼氣過程排出。
基于對上述過程的理解,開發出了ACI、NGI、MSLI等一系列模擬體內DPI沉降的儀器。
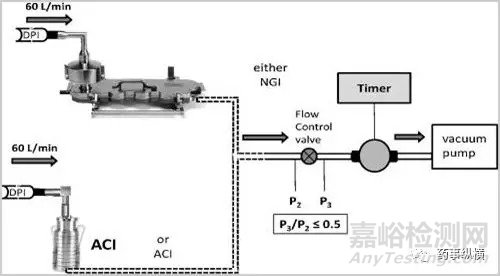
該類設備的工作原理簡述為在一定流速以及壓差條件下氣溶膠在不同孔徑的級段內發生撞擊并產生沉降進而區分出不同的空氣動力學粒度顆粒進而關聯體內吸收部位的沉降;基于設備的設計機理只能近似的模擬體內的吸入過程(例如制劑體內的吸入過程是一個在一定潮氣環境中的壓差遞減的吸入過程而上述設備常提供一個均衡的壓差);然而對于仿制制劑的開發而言該類設備提供了非常好的體外評價基礎。
DPI開發
基于對上述體內吸入過程的理解,吸入制劑的開發的重中之重則為如何實現適量藥物在肺內的沉積進而實現治療作用,而對于吸入仿制制劑的開發而言實現藥物在呼吸系統各部位的等量沉積是保證治療等效的重要基礎。顯然,吸入制劑的關鍵質量屬性除了常規制劑含量、有關物質等之外還包括APSD(AerodynamicParticle size Distribution)以及DDU(Delivered DoseUniformity)等其特有的質量屬性。吸入制劑終產品的獲得是一系列開發項目綜合作用的結果。主要影響因素包括配方、生產工藝、吸入裝置以及包裝密封系統,具體研究細節方面問題希望同行多多交流。
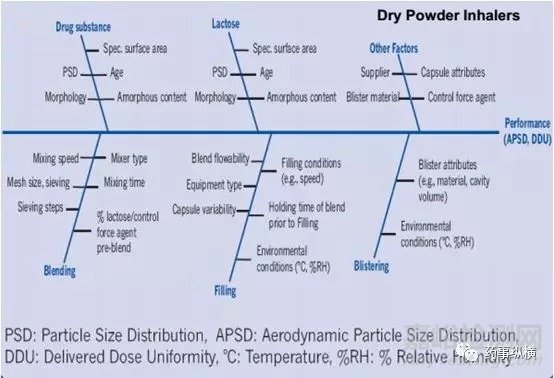
吸入制劑的開發涉及產品的多個維度,然最核心的維度此處筆者可簡述為:吸入制劑的開發的核心是對藥物分散、再吸附、再分散的過程理解;是對一系列力學平衡的過程的理解(主要包括原料與原料之間的內聚力以及原料與載體之間的粘附力的平衡,基于此平衡的達成會引入靜電力、范德華力、表面張力以及毛細管力等影響因素,故而任何配方開發、工藝開發、裝置開發以及包裝系統的開發都需以此為指導)。
本文多次提到空氣動力學粒度這一概念,眾所周知此概念并非指激光衍射法或顯微鏡方法測定的物理尺寸,實質上該粒度與物理尺寸和物理密度兩者綜合相關,故而在實際研發當中出現D90內控需在很小粒度之下時切勿大驚小怪。
國內外法規對比
我國15版藥典較10版藥典在吸入制劑的相關檢測方面有了較大的提高,然而與EP以及USP在相關項目的檢測方法和可接受標準上依然存在一定差異。主要對比如下:
中國藥典

歐洲藥典

美國藥典

生物等效性研究的要求
我國尚未頒布關于吸入制劑生物等效性方面的評價方法,下文簡要敘述EMA以及FDA對吸入制劑生物等效性方面的主要要求細則。EMA和FDA關于吸入制劑生物等效性研究方面各自持有不同的審評理念,這種差異主要體現為EMA認為體外評價的靈敏度高于體內評價故而當仿制產品達到體外APSD等的一致即可滿足等效性要求,FDA依然本著最科學的審評態度,生物等效必須建立在生物等效的研究之上。
EMA體外一致即可滿足生物等效性的具體要求:
1. 藥學一致(劑型、劑量、非活性組分)
2. 目標遞送劑量一致(T/R,±15%)
3. 對于DPI裝置吸入氣流阻力一致(T/R,±15%)
4. 吸入足夠量體積時確保活性物質肺內沉積量一致(T/R,±15%)
5. 每組或每級中的APSD分布一致(T/R,±15%)
若上述等效性不能建立則可進行肺內沉積相似性研究,沉積相似則等效性建立;若肺內沉積相似性不能建立則可進行臨床以及藥效學相似性研究,若建立則相似,若不能建立則“放棄治療”。
FDA生物等效性要求:
要求仿制藥需要證明與參比制劑具有相似的有效性和安全性,包括:裝置等效、體外等效、PK等效以及PD/TE等效。
筆者雜談
近年來FDA所批準DPIs大多是基于NDA進行批準,對于該類制劑的仿制開發已然成了眾多藥企的掘金之處。吸入制劑由于其較低的劑量以及對生產環境的高要求使得該類制劑開發具有較高的硬件要求,國內已有眾多企業在該劑型仿制領域內取得了一定的成績,筆者看來在該類劑型的仿制藥開發方面國內依然處于摸石頭過河階段,對于以創新型制劑開發的企業而言吸入制劑或可是一個較好的選擇。
然筆者亦處于摸索階段、經驗亦有限,文中不知之處還望同行批評指正。
參考資料
1.Regulatory Perspetives on Implementing QbD for MDIs and DPIs
2.Guidance for Industry Metered Dose Inhaler (MDI) and Dry Powder Inhaler (DPI) Drug Product document
3.CPMP points to consider on the requirements for clinical Documentation for orally Inhaled Product(OIP) CPMP/EWP/4151/00

來源:藥事縱橫


