您當前的位置:檢測資訊 > 生產(chǎn)品管
嘉峪檢測網(wǎng) 2025-08-02 21:14
摘要
目的:基于國家藥品抽檢工作,系統(tǒng)性評價風濕定膠囊的質(zhì)量以及分析存在的問題,為該品種的質(zhì)量控制和監(jiān)管提供參考。
方法:抽檢風濕定膠囊樣品136批次,進行標準檢驗,并開展探索性研究:采用高效液相色譜(HPLC)法建立制劑中白芷、徐長卿、甘草的含量測定和毒藜堿的限量測定,采用超高效液相色譜(UPLC)法建立風濕定膠囊特征圖譜,建立人工色譜的篩查方法,采用超高效液相色譜-質(zhì)譜聯(lián)用(UPLC-MS/MS)篩查風濕定膠囊中非法添加化學藥品的情況。
結(jié)果:136批次樣品根據(jù)現(xiàn)行質(zhì)量標準檢驗均合格,依據(jù)探索性研究進行評估,徐長卿、白芷、甘草含量測定存在低于擬定限度的情況,毒藜堿限量檢查未超出擬定限度,特征圖譜有部分企業(yè)的樣品出現(xiàn)缺失峰情況,部分樣品檢出人工色素及對乙酰氨基酚殘留。
結(jié)論:該次風濕定膠囊國家藥品抽檢的質(zhì)量狀況總體評價為“一般”。建議進一步完善質(zhì)量,重視原藥材質(zhì)量,加強生產(chǎn)企業(yè)自身管理,加強監(jiān)督檢查。
關(guān)鍵詞
風濕定膠囊;國藥藥品抽檢;質(zhì)量評價
國家藥品抽檢是通過構(gòu)建以問題導向的質(zhì)量分析體系,以探索性研究為核心手段,系統(tǒng)評估藥品質(zhì)量的穩(wěn)定性與標準可行性,識別潛在風險,為完善質(zhì)量標準、強化監(jiān)管效能提供科學依據(jù)。風濕定膠囊是國家藥品抽檢的中成藥品種之一,由八角楓、白芷、徐長卿、甘草4味藥材組成,具有活血通絡,除痹止痛之功效,用于風濕性關(guān)節(jié)炎,類風濕性關(guān)節(jié)炎,驚肋神經(jīng)痛,坐骨神經(jīng)痛。最早收載于部頒標準中藥成方制劑第十二冊,目前有十余份注冊標準,尚無藥典標準。
基于國家藥品抽檢以發(fā)現(xiàn)的問題為導向?qū)︼L濕定膠囊進行系統(tǒng)研究,結(jié)合前期調(diào)研工作、不良反應及多項探索性研究進行全面評估和綜合分析,發(fā)現(xiàn)了風濕定膠囊存在的質(zhì)量問題,現(xiàn)對其中關(guān)鍵探索性研究進行報道,以期對藥品監(jiān)督管理部門監(jiān)管提供建議,為其質(zhì)量標準提升提供理論依據(jù),保證藥品質(zhì)量更安全有效。
1儀器與試藥
1.1 儀器
Thermo Ultimate3000高效液相色譜儀,Agilent1260高效液相色譜儀,Waters Acquity UPLC超高效液相色譜儀,Xevo TQ-S三重四級桿質(zhì)譜儀;Waters Xbridge C18(250mm×4.6mm,5μm)色譜柱,Xselect HSS T3C18(250mm×4.6mm,5μm)色譜柱,ACQUITY UPLC®BEH C18(2.1mm×150mm×1.7µm)色譜柱,Waters HSS T3(2.1mm×100mm,1.8μm)色譜柱。瑞士梅特托利多AE200型電子天平(感量:0.01mg),XP26型電子天平(感量:0.01g);CAMAG薄層點樣儀。
1.2 藥品與試劑
甘草苷(批號:111610-201908,含量:98.0%)、甘草酸銨(批號:110731-202021,含量:96.2%)、丹皮酚(批號:110708-201908,含量:99.8%)、歐前胡素(批號:110826-201918,含量:99.0%)、異歐前胡素(批號:110827-201812,含量:99.6%)、檸檬黃(批號:510004-202003,含量:93.4%)、莧菜紅(批號:520002-201401,含量:87.4%)、胭脂紅(批號:111771-201302)、日落黃(批號:510005-201903,含量:90.9%)、亮藍(批號:112010-201501)、對乙酰氨基酚(批號:100018-201610,含量:99.9%)均來自中國食品藥品檢定研究院;毒藜堿(貨號:A637175,含量:98%)來自加拿大TRC公司。色譜甲醇、色譜乙腈來自天地公司,超純水,其余試劑均為分析純。
文章內(nèi)容由凡默谷小編查閱文獻選取,排版與編輯為原創(chuàng)。如轉(zhuǎn)載,請尊重勞動成果,注明【來源:凡默谷公眾號】。
2方法與結(jié)果
2.1 標準檢驗
本次抽檢共收到來自28個省(自治區(qū)、直轄市)136批次樣品,涉及生產(chǎn)企業(yè)12個,執(zhí)行標準9個,按各自標準檢驗,136批次樣品均符合規(guī)定。但由于執(zhí)行標準過多,部分標準過于簡單,項目設置不統(tǒng)一,不同企業(yè)之間、同企業(yè)不同批次之間,質(zhì)量差距較大。
2.2 探索性研究
以處方藥味及標準檢驗結(jié)果為基礎,根據(jù)調(diào)研情況以問題為導向,結(jié)合藥品質(zhì)量安全性和有效性的要求,開展了一系列探索性研究,結(jié)果顯示問題集中在風濕定膠囊中徐長卿、白芷、甘草的含量測定、特征圖譜、人工色素篩查和非法添加化學藥品檢查的項目中,現(xiàn)就這些項目進行具體闡述。
2.2.1 徐長卿、白芷的含量測定
鑒于徐長卿的主要活性成分丹皮酚屬酚類化合物,白芷中主要成分歐前胡素及異歐前胡素屬香豆素類化合物,兩者均具有極性較小的理化性質(zhì),適宜進行合并研究。本實驗對現(xiàn)行標準中方法缺失、指標等不統(tǒng)一等不足進行提升優(yōu)化。建立了高效液相色譜(high performance liquid chromatography,HPLC)法同時測定風濕定膠囊徐長卿中丹皮酚、白芷中歐前胡素和異歐前胡素含量方法,以期更好的控制風濕定膠囊中徐長卿、白芷的質(zhì)量。
采用Waters Xbridge C18(250mm×4.6mm,5μm)色譜柱,以乙腈-2%冰乙酸(50: 50)為流動相,丹皮酚檢測波長為274nm、歐前胡素和異歐前胡素檢測波長為250nm。對照品溶液的制備:取丹皮酚、歐前胡素、異歐前胡素對照品適量,精密稱定,加甲醇制成每1mL分別含丹皮酚15µg、歐前胡素10µg、異歐前胡素3µg的混合溶液,即得。供試品溶液的制備:取裝量差異項下的本品,研勻,取約1g,精密稱定,置具塞錐瓶中,精密加入甲醇100mL,稱定質(zhì)量,超聲(功率250W,頻率35kHz)處理60min,放冷,再稱定質(zhì)量,用甲醇補足減失的質(zhì)量,搖勻,濾過,取續(xù)濾液,即得。色譜圖見圖1。
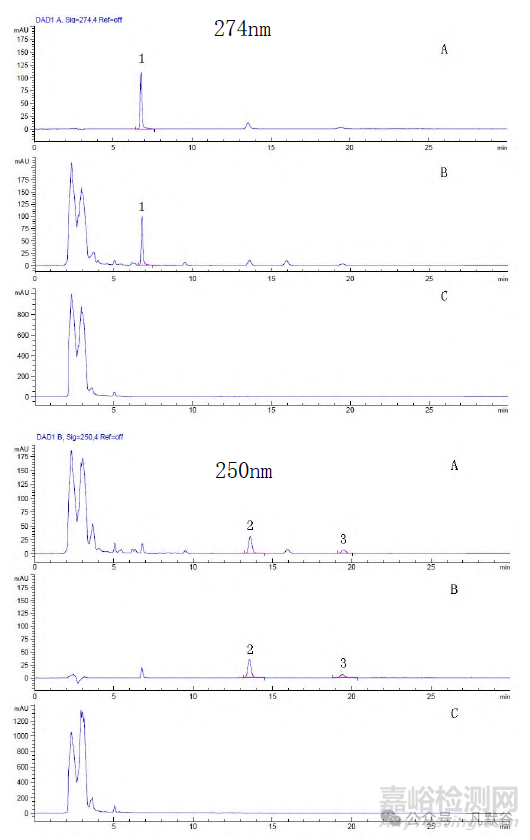
1.丹皮酚2.歐前胡素3.異歐前胡素。A. 陰性樣品;B. 混合對照品;C. 樣品。
圖1. 3 種溶液中徐長卿、白芷含量測定HPLC 色譜圖
1.paeonol 2. imperatorin 3.isoimperatorin. A. negative sample; B. mixed control; C. standard sample.
Fig.1 HPLC chromatograms for the determination of the content of cynanchim paniculatum and angelica dahurica in 3 solutions
歐前胡素和異歐前胡素為同分異構(gòu)體,根據(jù)測定值做相關(guān)分析,二者呈顯著的正相關(guān),鑒于2020年版《中華人民共和國藥典》白芷含量測定控制指標為歐前胡素,為使藥材與制劑控制指標一致,故以歐前胡素為指標進行控制和評價。根據(jù)制備工藝和實際測定值,擬定每粒含徐長卿以丹皮酚計不得少于0.35mg,每粒含白芷以歐前胡素計不得少于0.055mg。
結(jié)果部分批次丹皮酚低于擬定的限度。處方中徐長卿藥材9/10的量采用提取丹皮酚入藥,丹皮酚易揮發(fā),受溫度的影響較大,制劑工藝中的提取、干燥工藝均影響丹皮酚的含量。由圖2丹皮酚含量測定結(jié)果箱式圖可以看出,企業(yè)C、D、H部分批次丹皮酚的含量低于擬定的限度,與上述工藝參數(shù)控制有關(guān);企業(yè)F全部批次低于擬定的限度,提示該企業(yè)徐長卿的藥材原料質(zhì)量不佳。

圖2 丹皮酚含量測定結(jié)果箱式圖
Fig.2 Box plot of paeonol content determination results
部分批次歐前胡素低于擬定的限度。處方中白芷是生粉入藥,理論轉(zhuǎn)移率應為80%~90%或90%以上,但白芷中的香豆素成分受溫度影響較大,細粉的滅菌方式以及成品的干燥方式都可能對白芷的有效成分造成影響。由圖3歐前胡素含量測定結(jié)果箱式圖可以看出,企業(yè)B、D部分批次歐前胡素的含量低于擬定的限度,與上述工藝參數(shù)控制有關(guān);企業(yè)A、F、G、H全部批次低于擬定的限度,提示這些企業(yè)白芷的藥材原料存在質(zhì)量不佳或少投料問題。

圖3 歐前胡素含量測定結(jié)果箱式圖
Fig.3 Box plot of imperatorin content determination results
2.2.2 甘草的含量測定
本次國抽涉及的9份質(zhì)量標準均未對甘草的含量進行控制,本研究建立了風濕定膠囊甘草中甘草酸的含量測定方法,為控制和評價風濕定膠囊中甘草的質(zhì)量打下基礎。采用Xselect HSS T3-C18色譜柱(250mm×4.6mm,5μm)色譜柱,以乙腈(A)和0.05%磷酸(B)為流動相,進行梯度洗脫(0~12min,25%→43%A;12~18min,43%→60%A;18~19min,43%→60%A),檢測波長為237nm。對照品溶液的制備:取甘草酸銨對照品適量,精密稱定,加75%乙醇制成每1mL含甘草酸銨0.1mg的溶液,即得(甘草酸質(zhì)量=甘草酸銨/1.0207)。供試品溶液的制備:取裝量差異項下的本品適量,混勻研細,取約2.5g,精密稱定,置具塞錐瓶中,精密加入75%乙醇50mL,稱定質(zhì)量,加熱回流60min,放冷,再稱定質(zhì)量,用75%乙醇補足減失的質(zhì)量,搖勻,濾過,取續(xù)濾液,即得。色譜圖見圖4。

1.甘草酸銨。A. 陰性樣品;B.對照品;C. 樣品。
圖4. 3 種溶液中甘草含量測定HPLC 色譜圖
1.ammonium glycyrrhizinate. A.negative sample; B.control; C.standard sample.
Fig.4 HPLC chromatograms for the determination of glycyrrhizae radix content in 3 solutions
根據(jù)制備工藝轉(zhuǎn)移率和實際測定值,擬定每粒含甘草以甘草酸計不得少于0.17mg。多批次測定結(jié)果在擬定限度之下,包括企業(yè)A、E的全部批次,企業(yè)C、D、H的部分批次,測定結(jié)果的箱式圖見圖5。甘草的投料量甚小,故原料藥本身的質(zhì)量、準確的投料量、混合的均勻性可能是影響甘草質(zhì)量的較大因素。

圖5 甘草酸含量測定結(jié)果箱式圖
Fig.5 Box plot of glycyrrhizic acid content determination results
2.2.3 毒藜堿的限量檢查
八角楓中的毒藜堿(八角楓堿),為八角楓松弛肌肉的活性成分,也是主要毒性成分。本次抽檢有6份質(zhì)量標準對八角楓的生物堿進行了定量控制,測定方法有滴定法、薄層掃描法、高效液相色譜法,項目設置為檢查項、含量測定項。故須建立統(tǒng)一的、專屬性強、重現(xiàn)性好的測定方法,并制定合理的限度,以更好控制風濕定膠囊中毒藜堿的量。
采用Waters Xbridge C18(250mm×4.6mm,5μm)色譜柱,以甲醇-磷酸緩沖鹽(0.2g庚烷磺酸鈉,2g磷酸二氫鉀,0.3mL磷酸制成1000mL)(24∶76)為流動相,檢測波長259nm。對照品溶液的制備:取毒藜堿對照品適量,精密稱定,加氨水-甲醇(5∶95)制成每毫升含毒藜堿20µg的溶液,即得。供試品溶液的制備:取裝量差異項下的本品內(nèi)容物,研勻,取約1g,精密稱定,置具塞錐瓶中,精密加入氨水-甲醇(5∶95)50mL,稱定質(zhì)量,超聲(功率250W,頻率35kHz)處理30min,放冷,再稱定質(zhì)量,用氨水-甲醇(5∶95)補足減失的質(zhì)量,搖勻,濾過,取續(xù)濾液,即得。色譜圖見圖6。

1.毒藜堿。A. 陰性樣品;B.對照品;C. 樣品。
圖6 3 種溶液中毒藜堿測定HPLC 色譜圖
1.anabasine. A.negative sample; B.control; C.standard sample.
Fig.6 HPLC chromatograms for the determination of anabasine in 3 solutions
以標準檢驗測定的總生物堿數(shù)據(jù)與擬定方法測得的毒藜堿的數(shù)據(jù)做相關(guān)分析,p=0.007<0.05,不認為總生物堿與毒藜堿之間有線性關(guān)系,總生物堿與毒藜堿呈顯著的中等負相關(guān),見表1和圖7,即毒藜堿越高,總生物堿越低。各地方藥材標準未對八角楓進行定量,文獻報道鮮有提及毒藜堿的安全用量范圍,通過使用ADMET Predictor毒性預測軟件,未提供毒理風險系數(shù)評分和最大推薦治療劑量MaxRTD,故參考現(xiàn)行質(zhì)量標準限度,擬規(guī)定每粒含毒藜堿不得過0.35mg。136批次均在擬定限度內(nèi),說明各生產(chǎn)企業(yè)對風濕定膠囊中有毒成分毒藜堿的限度控制得較好,測定結(jié)果的箱式圖見圖8。
表1 總生物堿與毒藜堿相關(guān)分析
Tab.1 Correlation analysis between total alkaloids and anabasine mg·粒-1

圖7 總生物堿與毒藜堿含量關(guān)系
Fig.7 Relationship between total alkaloid and anabasine

圖8 毒藜堿測定結(jié)果箱式圖
Fig.8 Box plot of anabasine content determination results

2.2.4 風濕定膠囊的特征圖譜
采用UPLC建立高效快捷的風濕定膠囊特征圖譜評價方法,充分考慮制法中的生產(chǎn)工藝,以按制法制備的模擬小樣為參照制劑,考察制劑中4味藥材的投料質(zhì)量及按工藝生產(chǎn)情況。建立的特征圖譜共確定了22個峰的歸屬,11個特征峰指認到八角楓,6個特征峰指認到白芷,1個特征峰指認到徐長卿,4個特征峰指認到甘草,見圖9,經(jīng)方法學驗證可行,結(jié)合測定結(jié)果特征峰缺失情況與相似度進行評價。

6.甘草苷;11.丹皮酚;18.歐前胡素;20.異歐前胡素;21.甘草酸銨。
圖9 風濕定膠囊對照特征圖譜
6.glycyrrhizin; 11.paeonol; 18.imperatorin; 20.isoimperatorin; 21.ammonium glycyrrhizinate.
Fig.9 Reference characteristic chromatogram of Fengshiding capsules
結(jié)果顯示同時檢出22個特征峰且與模擬小樣相似度大于0.90的生產(chǎn)企業(yè)共有4家,分別為B、C、D、I企業(yè),說明這些企業(yè)依制法生產(chǎn),且使用的藥材成分較為穩(wěn)定。J、K企業(yè)同時檢出22個特征峰,但與模擬小樣的相似度僅為0.62和0.39,說明這些企業(yè)的樣品和模擬小樣之間存在顯著差異,可能是由于藥材來源的變化、提取工藝的不同、或者其他生產(chǎn)環(huán)節(jié)中的變異導致。A、E、L企業(yè)缺失八角楓和甘草的特征峰,F(xiàn)、G、H企業(yè)缺失八角楓、白芷、甘草的特征峰,說明這些企業(yè)投料的藥材質(zhì)量和生產(chǎn)工藝參數(shù)控制均存在較大問題。
2.2.5 人工色素的篩查
研究中發(fā)現(xiàn)K企業(yè)樣品的甲醇溶液均呈明顯的綠色,與其余11家企業(yè)的黃棕色溶液有明顯區(qū)別(圖10),故以發(fā)現(xiàn)產(chǎn)品顏色異常的問題為導向,對其“綠色”原因展開研究。采用顯微鑒別篩查葉類顯微組織、薄層色譜法篩查葉綠素,排除了“綠色”來源于葉類組織的可能性;隨后進行人工色素篩查,用聚酰胺固相萃取小柱富集色素,用高效液相色譜儀定量測定(圖11),并用液質(zhì)聯(lián)用進行確認(圖12),確定了K企業(yè)的所有批次樣品中均含有檸檬黃、莧菜紅、胭脂紅、日落黃、亮藍5種人工色素,含量分別為0.0979~0.1474、0.1190~0.4739、0.1831~0.3985、0.7763~1.3652、0.4407~0.5995mg·g-1。參考國標GB2760-2024《食品安全國家標準食品添加劑使用標準》的規(guī)定,食品添加劑使用時所允許的最大添加量:檸檬黃為0.5mg·g-1,莧菜紅為0.3mg·g-1,胭脂紅為0.5mg·g-1,日落黃為0.6mg·g-1,亮藍為0.5mg·g-1。K企業(yè)樣品的莧菜紅、日落黃、亮藍的檢出量超過最大添加量,屬于非法染色情況,具有較大的安全風險。
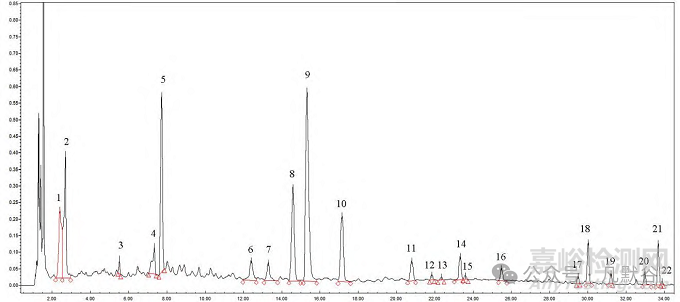
圖10 不同企業(yè)樣品甲醇溶液顏色比較
Fig.10 Color comparison of methanol solutions from different companies’samples

1.檸檬黃;2.莧菜紅;3.胭脂紅;4.日落黃;5.亮藍。A.混合對照;B.K 企業(yè)樣品; C.自制小樣。
圖11 人工色素HPLC 色譜圖
1.lemon yellow; 2.amaranth red; 3.carmine red; 4.sunset yellow; 5.brilliant blue. A.mixed control; B.sample from company K; C.self-made sample.
Fig.11 HPLC chromatogram of artificial pigments

1.檸檬黃;2.莧菜紅;3.胭脂紅;4.日落黃;5.亮藍。A.混合對照 B.K 企業(yè)樣品 C.自制小樣。
圖12 人工色素TIC 圖
1.lemon yellow; 2.amaranth red; 3.carmine red; 4.sunset yellow; 5.brilliant blue. A.mixed control; B.sample from company K; C.self-made sample.
Fig.12 TIC chart of artificial pigments

2.2.6 非法添加化學藥的檢查
中成藥中非法添加化學藥的現(xiàn)象時有發(fā)生,患者在不知情下長期服用此類“中成藥”,將產(chǎn)生不可預知的后果。本實驗采用UPLC-MS/MS的方法篩查風濕定膠囊中非法添解熱鎮(zhèn)痛類、抗風濕類等化學成分的情況,篩查了氨基比林、對乙酰氨基酚、保泰松、萘普生、吲哚美辛、奧沙普秦、雙氯芬酸鈉、尼美舒利、吡羅昔康、甲氧芐啶、醋酸伯尼松龍、地塞米松、氫化可的松13種化學物質(zhì)。結(jié)果C企業(yè)22批次樣品檢出對乙酰氨基酚,含量在8~32µg·g-1之間,屬于殘留量級別,排除故意添加化學藥的行為。
2.3 綜合評分與總體評價
根據(jù)檢驗項目與權(quán)重系數(shù)進行綜合評分。因現(xiàn)行標準不一致,按現(xiàn)行標準檢驗納入綜合評分的僅包含相同的檢驗項目,即水分、崩解時限、裝量差異3項,權(quán)重系數(shù)分別為1、0.9、0.8;按探索性檢驗納入綜合評分的檢驗項目有徐長卿、白芷、甘草的含量測定、毒藜堿限量檢查、人工色素檢查及非法添加化學藥物6項,權(quán)重系數(shù)分別為1、0.8、0.8、0.9、1、0.8。從圖13風濕定膠囊綜合評分結(jié)果圖可以看出,按現(xiàn)行標準檢驗進行評分,各企業(yè)的綜合評分接近,無法全面反映風濕定膠囊的質(zhì)量狀況;按探索性項目評分后,各生產(chǎn)企業(yè)的綜合評分差異顯著,反映出在風濕定膠囊生產(chǎn)中存在潛在的質(zhì)量問題。本次風濕定膠囊國家藥品抽檢的質(zhì)量狀況總體評價為“一般”。
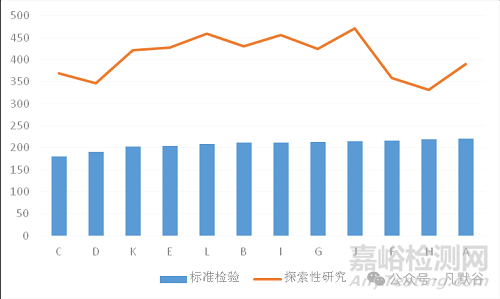
圖13 風濕定膠囊綜合評分結(jié)果
Fig.13 Result of comprehensive scoring for Fengshiding capsules
3討論
3.1 發(fā)現(xiàn)的問題
質(zhì)量標準缺失引起的投料藥材質(zhì)量控制問題:本次探索性研究統(tǒng)一了徐長卿、白芷、甘草的含量測定方法,暴露出因質(zhì)量標準項目的缺失引發(fā)的各藥味的質(zhì)量控制問題,原標準有含量測定項目的測定結(jié)果較好,沒有的則測定數(shù)據(jù)普遍較低,提示質(zhì)量標準對于企業(yè)投料生產(chǎn)具有重要的指導性意義,同時企業(yè)還需注意細化生產(chǎn)工藝、干燥方式等。
非法染色問題:近年來,一些不法生產(chǎn)企業(yè)通過非法染色來掩蓋藥品質(zhì)量問題的情況時有發(fā)生,人工色素大多含有偶氮和苯環(huán)結(jié)構(gòu),這些成分經(jīng)過人體代謝后可能會產(chǎn)生致癌或致畸的物質(zhì),對健康構(gòu)成威脅。通過本次探索性研究,K企業(yè)生產(chǎn)的風濕定膠囊樣品中檢測出五種人工色素,其含量超過或接近推薦的最大添加量,暴露出該企業(yè)原輔料采購環(huán)節(jié)的質(zhì)量控制存在明顯缺陷,可能采購了非法染色的中藥材或顏色穩(wěn)定性不達標的輔料,進而引發(fā)了非法染色問題。提示K企業(yè)在風濕定膠囊生產(chǎn)過程中,針對非法染色的質(zhì)量控制管理存在嚴重漏洞。
化學藥殘留問題:國家明令禁止在中成藥中非法添加化學藥,但在C企業(yè)生產(chǎn)的風濕定膠囊中檢測出了對乙酰氨基酚的殘留。經(jīng)國家藥品監(jiān)督管理局網(wǎng)站數(shù)據(jù)庫檢索,C企業(yè)也生產(chǎn)含有對乙酰氨基酚的化學藥品,提示該企業(yè)共線生產(chǎn)設備清場過程存在漏洞,清潔驗證體系不完善,其應對藥物殘留應格外關(guān)注,避免更大的交叉污染。
3.2 建議
應進一步完善質(zhì)量標準,增加對關(guān)鍵藥材有效成分含量的測定,并強化對非法添加化學藥品和人工色素的篩查,以確保產(chǎn)品的安全性和有效性。政府需加強對中藥材市場的監(jiān)管,打擊制假制劣及非法添加行為,同時企業(yè)應強化中藥材及飲片的專業(yè)鑒定和入庫檢驗,確保所用藥材的質(zhì)量符合要求。生產(chǎn)企業(yè)應建立完善的質(zhì)量管理體系和內(nèi)部監(jiān)督機制,提高半成品的控制指標,增加有效成分和限量成分的監(jiān)控,并重視共線生產(chǎn)的清潔驗證,防止交叉污染。監(jiān)督部門應定期對生產(chǎn)現(xiàn)場、原材料和成品進行抽樣檢測,對存在問題的企業(yè)進行查處并督促其整改到位。

來源:Internet


