分享一下Lufotrelvir工藝研究的文獻
最初路線
化合物3,先制備氯代酮4,去Boc得到化合物5,構建酰胺鍵得到化合物6,再脫Boc得到化合物7
化合物7和化合物9,經酸胺縮合得到化合物8
化合物8,和帶拉電子基團的羧酸鈉反應,然后水解引入羥基得到化合物1,再引入磷酸酯基團得到化合物2。
化合物9的制備,安全性不好
化合物3制備化合物4,放大重現性不好
化合物2因為工藝問題,穩定性不好。
多步中間體存在穩定性問題,控制點不夠。
改進路線
思路:改變片段的拼接順序,最后引入化合物3片段,更多的結晶分離點,易于質量控制。
吲哚環酰胺片段13的研究
路線A
化合物9A和草酸二乙酯,先叔丁醇鉀條件發生親核取代,再水解反應,雷尼鎳還原硝基后,酸處理關環得到吲哚環羧酸9
再和化合物14進行酸胺縮合得到化合物15,堿的選擇是關鍵,三乙胺也會導致消旋,吡啶或者N-甲基咪唑消旋風險低。
水解甲酯,同樣不能選擇堿性,采用酸性水解,溫度不能高,高了會導致甲醚鍵斷裂。
路線B
路線A路線可行,但是供應商不能穩定供貨,開發了路線B
化合物9C在LDA條件下與DMF作用引入甲酰基9D,再銅催化關吲哚環得到化合物9E
反應的關鍵是Ullmann反應引入甲氧基,配體很關鍵,研究發現草酸酰胺的配體效果更有效,DEPO或者DMAPO。
內酰胺片段5的研究
最初路線采用氯碘甲烷構建氯代酮,放大重現性不好
改用氯乙酸法,采用傳統反應釜LiHMDS和叔丁基氯化鎂都可行,但是改用流動反應,因為鋰鹽溶解度的問題,反應不如叔丁基氯化鎂好。
采用對甲苯磺酸去除Boc可得固體,鹽酸或者甲基磺酸只能得到油。
酰胺鍵8的構建研究
化合物13和化合物5制備化合物8
化合物13堿性環境容易消旋
最初條件采用HATU,-20度下,滴加DIPEA,10-20g規模可接受,放大到500g消旋嚴重,和DIPEA滴加速率導致體系pH值有關。
繼續研究采用Zn(OTf)2 和EDCI組合,可以在一定程度抑制消旋,放大到600g,供毒理批物料需求。
工藝的繼續研究
雖然采用采用Zn(OTf)2試劑可以抑制消旋雜質,但是化合物5自身的各種二聚雜質很難控制,導致收率很低。
調整順序,改進策略
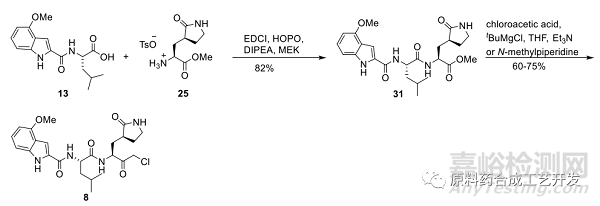
采用化合物25為底物,先和化合物13進行酸胺縮合,再制備氯代酮。
采用MEK或者丙酮為溶劑,采用EDCI和HOPO組合,DIPEA為堿,反應結果想對最好。
化合物31質量很好,可直接用于下一步制備氯代酮化合物8
氯代酮8的制備研究
篩選
硫葉立德法和氯乙酸或者氯乙酸酯法,對比后選擇氯乙酸/叔丁基氯化鎂法
研究
底物結構中含有四個質子,需要消耗額外的叔丁基氯化鎂,當量需求大。
大過量叔丁基氯化鎂的使用有助于脫氯雜質的產生
初步研究加三乙胺可以抑制脫氯雜質,進一步研究發現有親核性的N-methylpiperidine或者N-methylpyrrolidine更有助于抑制脫氯雜質。.
加料順序
底物31和氯乙酸緩慢滴加到叔丁基氯化鎂和N-甲基哌啶的THF中,有助于雜質控制
化合物31氯乙酸/叔丁基氯化鎂制備氯代酮,反應過程類似依托考昔的某步反應
制備API的后續路線
參考文獻:https://doi.org/10.1021/acs.oprd.2c00375