您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2022-08-18 12:19
新藥開發以普通口服固體制劑居多,制劑經過胃腸道吸收進入體循環,早期制劑以獲得足夠的體內暴露為目標,以便研究新的化合物分子在體內的藥理學,藥代動力學和毒理學情況。根據這幾年從事新藥開發的狀況來看,制劑人員更多是被動接受新藥藥物分子結構以及確定的固態開發形式,在前期藥物發現階段,參與的深度還是較低的,當然每個公司的開發策略與模式是不同,而且其中還涉及項目的保密。但是,萬變不離其宗,拿到一個新的化合物,還是要從性質入手,包括其結構特征,理化性質,生物學性質以及PK,毒性等。在新藥開發早期,API的可及性往往限制研發的推進,如何在化合物眾多理化性質中把握關鍵性質,抓住主要矛盾,將是我們制劑開發人員需要思考的問題。
生物藥劑學分類系統(BCS)的橫空出世,似乎給了我們指引關于新藥早期開發需要格外關注的藥物性質-溶解度和滲透性,其背后的關鍵性質不僅是獲得體內生物等效性研究豁免的有用工具,還是FDA建議的新藥發現和早期開發決策的有用工具。當然,除了BCS所要關注的藥物性質之外,化合物的穩定性在藥品開發中也是需要足夠關注。預計通過幾篇小的文章,給大家介紹一下,新藥早期開發中需要關注的關鍵的藥物性質,包括溶解度,滲透性以及穩定性。本文主要介紹一下有關藥物溶解度的相關信息。
對于溶解度的認識確實是不斷的發展與深入,而且不同的書籍與著作可能采用不同的觀點與方向去講訴有關溶解度的故事,隨著學習的加深,確實也獲益匪淺,一定程度上,避免了一直被領導支配的恐怖,他可能上嘴唇碰著下嘴唇,下面的人就要跑斷腿。說的有點夸張,確實也看到有的同事反復去執行領導的要求,也做了很多次平衡溶解度。對于固體制劑開發怎么重視都不為過,但是,是否需要反復去確認呢?
以下幾個問題供大家思考:
是否可以通過其他的性質間接去證明溶解度未發生變化?
多晶型影響溶解度嗎?
粒徑及粒度分布改變了是否需要重新研究平衡溶解度?
非pH依賴的藥物分子如何設計各介質條件?
怎么確定平衡溶解度達到平衡,還是所有化合物24h即可?
以上問題或許無法在本文找到答案,可以看看本人歷史上的小文,或者借助工具書,去探索究竟。下面我將開始正式開始本文的進程(以下提高的溶解度概念皆指平衡溶解度,除非特別說明)。
基本概念:溶解系指一種或一種以上物質以分子或離子狀態分散在另一種物質中形成均勻分散體系的過程,溶液中任意部分都具有完全一致的性質。在溶解的物質(即溶質)分子與分散介質(即溶劑)分子產生相互作用時,如果不同種分子間的相互作用力大于同種分子間作用力,則溶質分子從溶質上脫離,繼而發生擴散,最終在溶劑中達到平衡狀態,即溶質的溶解速度與其結晶速度相等。所以也可以說,物質的溶解是溶質和溶劑的分子或離子相互作用的過程,這種相互作用力主要是范德華力、氫鍵力和偶極力。
以上說法,其實很多書和我以前寫的小文都提到過,下面的圖1不少文章也有,再次去介紹關于平衡溶解度的概念,其實還是想重申平衡的含義:溶質脫離晶體結構中的溶解速率與溶劑中溶質的析出達到了動態的平衡。這樣就要求我們,一來我們做平衡溶解度試驗的時候需要根據化合物的大概溶解度情況(預實驗或者計算的方式),抑或著加入大量API于介質中(新藥早期開始對于溶解度比較好的化合物還是需要不少量的),保證平衡時溶質仍存在未溶解的部分。二來對于什么時間達到平衡,不同的化合物且相同的化合物不同的介質環境應該都是有所差異。有的化合物24h,48h甚至72h。如何證明24h達到平衡,起碼你需要再檢測24h以后的一個時間點,證明后一個時間點與24h溶解度無明顯差異。至于如何說明這個無明顯差異,這個尺度如何把控,暫未發現報道。三來平衡是溶液所具有的濃度為該化合物的飽和溶解度。無論化合物成鹽,轉變為無定形或者亞穩定性態,其平衡溶解度應該都是一致的。
有必須說明一下,不同固態形式可能可以提高溶解度,但是也是有條件要求的。比如化合物成鹽提高溶出速率,形成過飽和溶液,看似溶解度提高了,可是如果在特定pH介質且緩沖能力足夠強,就可以消除成鹽反離子的作用,化合物無論成鹽,還是游離堿,游離酸,溶解度是相同的;對于化合物無定形來說,看似也是提高了溶解度,無定形固體分散體以極小的粒度,小到可以達到分子級別,而且無定形的API需要親水性載體來穩定,這樣也提高ASD的潤濕性,而且無定形本身就是打破了溶解所需要的晶格的限制,破除了高熔點固態化合物溶解的限速步驟,這樣提高最終制備的ASD中藥物的溶解度似乎板上釘釘。但是,首先需要明確一定,ASD其實是不具有平衡溶解度,因為無定形態固有的結晶趨勢,因此實際測量的溶解度可能無法達到最大可達到的無定形平衡溶解度。為了確定無定形態到底能提高把API的溶解度提高到了多少,可以通過理論公式去計算。

ASD開發化合物的溶解度理論上增強可以是幾個數量級,大多數藥物溶解度可以提高2-20倍,對于極高熔點的,甚至可以提高50倍。然而,當將幾種無定形藥物的預測溶解度值與文獻中的實驗報告值進行比較時,通常存在較差的相關性。這種不一致部分歸因于無定形態物質快速結晶成亞穩定性態,接著又會由亞穩態向穩定態轉變,最終溶液濃度和穩定型晶型的平衡溶解度一致。通過上面的解釋,這樣我們就可以理解,物理藥劑學上對于溶解度的定義:藥物在溶劑中的熱力學溶解度是指一定溫度和壓力的平衡條件下,在一定體積的溶液中所含有的最穩定晶型藥物的最大值。熱力學溶解度一般指平衡溶解度。

圖1.溶質藥物分子從固體顆粒中置換出來并進入溶液需要三個基本步驟。第一步:從晶格中去除單個溶質分子;在此步驟中需要能量來克服固態中的溶質-溶質相互作用。第2步:在溶劑中形成空隙以容納溶質分子。盡管此步驟也需要能量,但它可能比步驟1所需的能量低得多。步驟3:溶質分子插入溶劑中,形成溶質-溶劑相互作用。簡單地說,如果溶質-溶劑相互作用(即第3步)釋放的能量大于第1步和第2步所需的能量,則對于溶解度是有利的。
溶解度的限速步驟:由圖1可知,藥物溶解度有兩個主要決定因素:1)克服溶質固態分子間力強度所需的能量和2)能量溶液中溶質和溶劑分子相互作用產生的(溶劑化)。
根據一般溶解度方程(GSE),可以估算藥物在水中的固有溶解度(S0):
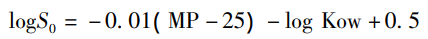
MP為熔點(℃),Kow為油水分配系數。其實,這個方程,也介紹不少次了,該方程很好的契合了圖1中所描述的一個晶體藥物的溶解過程,提取了兩個重要的可能限制溶解度的參數,即晶型藥物的熔點和logP。當然,同一化合物不同的晶型之間熔點是不同,其可以表征固態形式的差異,是反映化合物固態形式的固有屬性的一個參數。LogP反應化合物親脂性的參數,更加強調分子結構的特性,與固態形式無關,即同一分子LogP是相同的。這樣也很好解釋上述提出的一個問題:多晶型影響溶解度嗎?
多晶型之間熔點的差異,LogP一致,帶入上述公式,是不是表明其溶解度發生變化了。那么問題來了,合成可能合成很多批次的原料,晶型不變,還需要反復去驗證溶解度是否發生變化嗎?
其實,從上面的計算公式,我們還可以推出新的結論。當溶解度數值發生1個對數單位變化,LogP也會相應的變化一個對數單位。而熔點需要變化100℃的情況下,溶解度才發生1個對數單位變化,可見LogP對于溶解度的影響可能遠遠大于熔點。這樣也解釋了一般認為200多℃為高熔點,但是仍有化合物溶解度可以達到200μg/ml以上的溶解度;而熔點100多℃,仍舊可能是難溶性藥物。
最后還要補充一點的是,有的文獻/書籍中說化合物的溶解度是其化合物的固有屬性,固有屬性可以理解為在條件一定下,其溶解度應該是確定不變的。可是,有的資料又說,化合物溶解度并沒有單一的數值。一開始感覺這兩份資料,是不是有點矛盾。其實,在藥典里面,很少去報道該化合物具體的溶解度數值,因為化合物的溶解度的數值的測定具有不確定性,影響因素太多了,包括化合物分子結構本身的屬性,固態形式,固態形式純凈程度,配制的介質,試驗操作(離心VS過濾膜,離心多久,會不會因為離心過程溫度變化結晶析出)等等。從理論上說,控制這些變量,理論上在一定條件下,溶解度是確定的。可是在真實的世界,平衡溶解度的數值又很難去統一。當然,即使有些條件無法全部統一可能造成結果有偏差,但不可能具有質的變異,除非你的試驗出現了重大的失誤,設計具有問題。
小結:新藥的開發,項目是無比的急切地,可能在實際研發中多次變更整個IND申報的試驗計劃,輪到制劑這邊往往,計劃可以推遲,但是計劃的終點依舊不變。如何在所剩不多的時間中,掌握主動權,即使痛,還要快樂著(強顏歡笑),那就需要我們不斷地武裝自己,在平時夯實基本功,提高對制劑開發中的相關知識掌握的深度,形成自我的知識體系。
最后絮叨一句。以上除了一些書本和文獻中的內容,還加入了不少的個人的理解與感悟,如有不妥,還請及時反饋,以免給予大家以誤導!
參考文獻:
1.Strategies to Address Low Drug Solubility in Discovery and Development
2.Martin物理藥劑學與藥學。(書籍)
3. Amorphous solid dispersions: Utilization and challenges in preclinical drug development within AstraZeneca
4. Pharmaceutical Amorphous Solid Dispersions (書籍)

來源:藥事縱橫


