在選擇何種晶型定量研究方法前,我們需要先了解下不同方法的優缺點以及適用的情形,結合所研究的藥物特性,開發準確的可用于晶型定量檢測分析的方法,來保證藥物質量的一致性、穩定性、安全性及有效性。
為什么要進行藥物晶型定量?
同一藥物的不同晶型可能導致藥物的穩定性、生物利用度及療效發生改變,因此對于藥品質量與臨床療效都有影響,對藥物多晶型的研究,尤其在晶型定量控制方面頗為重要。
國家藥物監管機構對多晶型藥物定量也有明確要求。2020年版《中國藥典》提到當固體藥物存在多晶型現象,且不同晶型狀態對藥品的有效性、安全性或質量可產生影響時,應對原料藥物、固體制劑、半固體制劑、混懸劑等中的藥用晶型物質狀態進行定性或定量控制。
不同晶型定量方法差異
常用的定量分析方法有單晶X射線衍射法(SXRD)、粉末X射線衍射法(PXRD)、差示掃描量熱法(DSC)、紅外光譜法(IR)等。在藥物研究中也有多個實用案例,如利用X射線衍射單峰法控制原料藥中的雜質含量,以特征峰的峰高對含量作線性回歸,計算得樣本實驗值與真實值線性回歸的相關系數;利用X射線衍射結合偏最小二乘法(PLS)法進行制劑中的定量分析研究等。
以下為常用的晶型定量方法的優缺點
|
方法 |
優點 |
缺點 |
|
粉末X射線衍射法(PXRD) |
單峰法 |
方法建模簡單靈敏度也相對較高 |
嚴重依賴于標樣的純度制樣擇優取向效應嚴格控制操作條件 |
|
全譜法 |
準確度高 |
方法建立和計算過程非常復雜 |
|
差示掃描量熱法(DSC) |
方法操作簡單,試樣用量少 |
適用范圍局限,僅適用于不同晶型物質的熔融吸熱峰存在較大差異或供試品中含有不同數量和種類結晶溶劑的晶型物質 |
|
紅外光譜(IR) |
可以獲得較高的光譜分辨率、信噪比和準確性 |
易受樣品中雜質的干擾影響,常作為PXRD輔助方法 |
|
動態蒸汽吸附法(DVS) |
樣品用量少,可對無定型進行定量 |
適用范圍局限,適用于樣品在特定濕度時有晶型轉變,且吸水量有明顯變化的體系 |
PXRD技術晶型定量優勢
藥物晶型分析貫穿原料藥研究與開發、制劑研究與開發等藥物質量控制的各個階段,與其他方法相比,PXRD是最經典、非破壞性、較準確的晶型定量方法,且PXRD現已成為藥物晶型的定量分析中的主要通用方法。且隨著儀器、配件及軟件的不斷升級及發展,PXRD可以實現更快、更靈敏、更高通量的分析。
PXRD準確度高,分辨能力強,可通過給出晶胞參數(如原子間距離、雙面夾角等)確定藥物晶型與結構,可用于鑒別晶態與非晶態、混合物與化合物的定性分析。在晶型定量應用中,每種相的各衍射線條d值(晶面間距)和雙面夾角也不變,但混合物中各物相之間的相對強度會隨各相在混合物中的百分比含量而變化。Alexander等于1948年首次提出了用于粉末混合物定量的基本參數,并推導了峰強度與樣品中相應組分晶型含量的數學關系式,這奠定了該方法用于定量分析的理論基礎。
此外,PXRD的定量分析方法又包括單峰法和全譜擬合法。單峰法相對簡單,對樣品的信息要求較低,靈敏度也相對較高,是常用的一種定量分析方法。但嚴重依賴于標樣的純度制樣擇優取向效應,需嚴格控制操作條件。全譜法是以化學計量學為基礎,如偏最小二乘法、全粉末圖譜分解法、Rietveld法等方法對樣品進行定量分析,其信噪比、靈敏度和專屬性均比單峰法有明顯提高。但是方法建立和計算過程非常復雜,對樣品圖譜的解析能力要求也較高,所以實際應用相對較少。
PXRD晶型定量中存在的問題
PXRD的擇優取向是導致定量結果偏差的重要因素。在多晶型中,各晶粒的取向向某些方位偏聚,導致衍射峰強度異常,從而對定量結果產生負面影響引起偏差。
經驗表明,以下方法可以降低擇優取向對PXRD檢測結果的影響:
降低樣品的顆粒尺寸:通過減小樣品的顆粒尺寸,可以減小各晶粒的取向差異,從而降低擇優取向的影響。這種方法適用于難以獲得微細粉末的樣品。
旋轉樣品臺:在PXRD實驗中,通過旋轉樣品臺可以使各晶粒的取向分布更加均勻,從而降低擇優取向的影響。這種方法需要在樣品和掃描參數的選擇上加以注意。
選擇合適的步長和步速:通過選擇合適的步長和步速,可以改善譜峰之間的分離度,從而減小擇優取向的影響。
總的來說,在使用PXRD進行晶型定量時,應該根據具體的實驗條件和要求采用不同的測量程序。例如,可以通過更加細致的樣品處理方法改善數據統計模型的準確性;通過更長的信號采集時間提高數據的質量;通過更合理的傳輸參數優化實驗條件等。這些方法都可以幫助降低擇優取向對PXRD檢測結果的影響,從而提高定量結果的準確性。
雜質晶型定量案例
1、案例背景
該產品的制作工藝較為復雜,其中主要離子為硬路易斯酸,易與氫氧根離子結合,制備時需要控制pH值在酸性條件下,將氯化物溶液倒入碳酸氫鈉溶液中形成晶核,再以晶核為中心逐漸擴大形成結晶,其中以4個結晶水的結晶生物利用度最高,藥效最好。若制備過程中反應條件控制不當會導致目標藥物中混有雜質晶型,而美國FDA從未批準其雜質晶型用于適應癥的臨床治療。
目的:
使用PXRD對樣品的不同雜質晶型進行表征,建立相應的定量分析方法。
步驟:
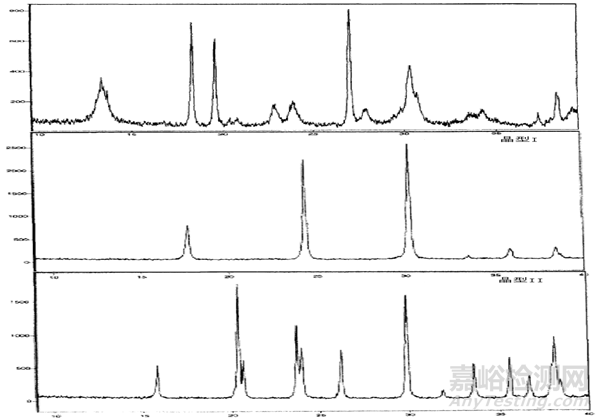
定性分析:首先,通過對藥物及其兩種雜質晶型的X射線衍射圖譜進行比較,可以確定不同晶型的存在。
建立定量分析方法:
采用單峰法在X射線衍射的定量分析中,通常每個物相我們選取一條特征衍射峰來采集數據。選擇的衍射峰和其它物相的衍射峰不可發生重疊,盡可能選擇獨立且強度較高的特征峰。
a. 峰高法:選取每個晶型的特征峰,計算峰高強度比,得出目標晶型中雜質晶型含量的定量標準曲線。
b. 峰面積法:選取每個晶型的特征峰,計算峰面積強度比,同樣得出目標晶型中雜質晶型含量的定量標準曲線。
制樣注意事項:
a. 避免擇優取向:在制樣時,要特別注意避免樣品出現擇優取向的現象,以免造成強度不穩定、出現偏差。
b. 力度適中:如果采用的是干法研磨的手段,在研磨混合時力度要適中,重在混合而非用力研磨,否則可能會導致樣品粘附在研缽壁或研磨棒上,造成數據不準確、偏離真實值,甚至破壞晶型。
c. 少量多次加入雜質樣品:由于雜質晶型的含量較低,應一邊研磨混合一邊少量多次加入,這樣可以避免雜質樣品結塊,集中在同一區域從而造成較大實驗誤差。
本實驗在對主晶型及其兩種雜質晶型定性分析的基礎上,建立了基于PXRD單峰法中峰高法和峰面積法的定量分析方法。通過選取3種晶型的特征峰來計算峰高強度比和峰面積強度比,分別得出了兩種雜質在目標晶型中含量的定量標準曲線,兩者都有良好的線性關系,可以用于實際樣品的定量分析。