您當前的位置:檢測資訊 > 法規標準
嘉峪檢測網 2023-07-17 09:16
|
條款 |
要求 |
|
247 |
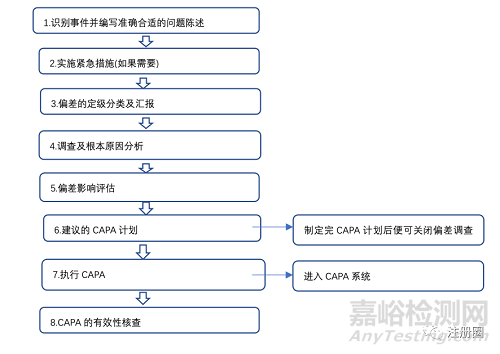
各部門負責人應當確保所有人員正確執行生產工藝、質量標準、 檢驗方法和操作規程,防止偏差的產生 |
|
248 |
企業應當建立偏差處理的操作規程,規定偏差的報告、記錄、調 查、處理以及所采取的糾正措施,并有相應的記錄。 |
|
249 |
何偏差都應當評估其對產品質量的潛在影響。企業可以根據偏 差的性質、范圍、對產品質量潛在影響的程度將偏差分類(如重大、次要偏差),對重大偏差的評估還應當考慮是否需要對產品進行額外的檢驗以及對產品有效期的影響,必要時,應當對涉及重大偏差的產品進行穩定性考察 |
|
250 |
何偏離生產工藝、物料平衡限度、質量標準、檢驗方法、操作規 程等的情況均應當有記錄,并立即報告主管人員及質量管理部門,應當有清楚的說明,重大偏差應當由質量管理部門會同其他部門進行徹底調查,并有調查報告。偏差調查報告應當由質量管理部門的指定人員審核并簽字。企業還應當采取預防措施有效防止類似偏差的再次發生。 |
|
251 |
量管理部門應當負責偏差的分類,保存偏差調查、處理的文件 和記錄 |
|
252 |
業應當建立糾正措施和預防措施系統,對投訴、召回、偏差、 自檢或外部檢查結果、工藝性能和質量監測趨勢等進行調查并采取糾正和預防措施。調 查的深度和形式應當與風險的級別相適應。糾正措施和預防措施系統應當能夠增進對產 品和工藝的理解,改進產品和工藝 |
|
253 |
企業應當建立實施糾正和預防措施的操作規程,內容至少包括: (一)對投訴、召回、偏差、自檢或外部檢查結果、工藝性能和質量監測趨勢以及其他來源的質量數據進行分析,確定已有和潛在的質量問題。必要時,應當采用適當的統計學方法; (二)調查與產品、工藝和質量保證系統有關的原因; (三)確定所需采取的糾正和預防措施,防止問題的再次發生; (四)評估糾正和預防措施的合理性、有效性和充分性; (五)對實施糾正和預防措施過程中所有發生的變更應當予以記錄; (六)確保相關信息已傳遞到質量受權人和預防問題再次發生的直接負責人; (七)確保相關信息及其糾正和預防措施已通過高層管理人員的評審。 |
|
254 |
施糾正和預防措施應當有文件記錄,并由質量管理部門保存。 |
|
偏差/事件開啟 |
|
|
偏差或事件 |
□偏差 □事件 |
|
發現部門 |
|
|
發現事件和日期 |
|
|
發現地點 |
|
|
發生時間和日期 |
|
|
發生地點 |
|
|
通知QA的日期/時間 |
|
|
偏差/事件的客觀描述包括涉及到的產品、批次、設備編號,所違背的SOP規定等 2015.04.23 11:36 實驗員汪某在做A1原液(批號為:A1201504018B-A1201504023B)卵清蛋白含量測定(酶聯法)時,內部標準品結果為12.0ng/m,公司內部SOP規定的內部標準品7.5ng/ml計算結果應在5-10ng/ml之間,不符合SOP規本次實驗無效。 |
|
|
立即采取的緊急措施(如有) 見第2步 |
|
2.實施緊急措施(如果需要)
|
偏差級別 |
定義 |
|
Critical deviation(關鍵偏差) |
對已上市的產品安全、鑒別、規格、質量、純度、效力、性能、可靠性、藥效持續時間等有潛在負面影響的偏差,或對臨床研究有潛在負面影響的偏差,或導致一個質量系統重大失敗的偏差。 |
|
Major deviation(主要偏差) |
對未上市或者用于臨床研究的產品安全性、鑒別、規格、質量、純度、效力、性能、可靠性、藥效持續時間等有一定影響的潛在的負面影響的偏差。這類偏差顯示的系統性失敗影響有限,或偏差在現有程序檢查時發現并可能再次發生。 |
|
Minor deviation(微小偏差) |
對產品安全、鑒別、規格、質量、純度、效力、性能、可靠性、藥效持續時間等沒有或者只有很小的負面影響的偏差,同時不偏離法規要求或者承諾。 |
附件A中提供了偏差定級的具體舉例。
|
偏差/事件編號 |
到期日期 |
||||||||||||||||||||||||||||||||
|
偏差/事件名稱
|
|||||||||||||||||||||||||||||||||
|
偏差調查信息 |
|||||||||||||||||||||||||||||||||
|
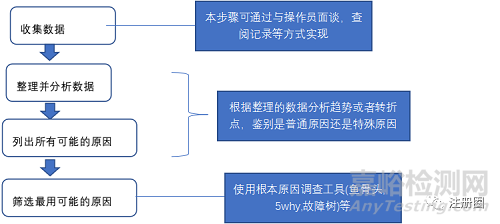
(1)對實驗員的調查 通過與實驗員面談,確認實驗過程中樣品加樣順序及額外等待時間與檢測SOP及試劑盒說明書不完全一致,但是這兩處的不一致并不影響產品檢測結果,因此不會影響內參結果,并不是偏差的根本原因。同時實驗員指出,卵清蛋白含量檢測標準曲線的相關系數R2比較低,通常僅有0.95左右,具體見下表:
通過進一步查看卵清蛋白含量檢測過程中的標準曲線,發現該曲線并不是并不是標準的一階回歸曲線,而是類似與拋物線的曲線,見下圖:
(2)對試劑盒供應商及gsk的咨詢 通過對試劑盒供應商的咨詢,廠家建議選擇四元回歸曲線。但目前公司QC的酶標儀不具備此功能。查閱gsk意大利工廠的卵清含量檢測Sop發現,意大利工廠采用二階回歸曲線來作為標準曲線,即Y=aX2+bX+C,其中a,b,c為常數,Y為卵清蛋白,X為對應的吸光度。同天咨詢公司的數理統計專家得到如下反饋:四參數模型可以使用較大范圍的ELISA實驗,例如卵清蛋白含量范圍從1ng/ml到1ug/ml范圍,由于胡可效應,標準曲線呈現隨待測樣品濃度增加而吸光度增加幅度越來越小。如下圖:
但在待測樣品濃度范圍較小時,可以使用一階回歸或者二階回歸來你和標準曲線,只要其方法誤差在允許范圍內即可。 卵清蛋白含量檢測分析方法標準曲線類型的確定:公司之前使用一階線性回歸曲線作為標準曲線,只要其方法誤差在可接受范圍內,方法就并非錯誤。內參作為實驗有效性的判斷工具,其真實含量為7.5ng/ml,可接受標準為5-10ng/ml,即標準的回收率為66.7%-133.3%。根據《分析方法驗證管理規程》,對于生物學檢驗,其準確性需要通過數據統計分析確定,因為該類方法不如儀器分析方法那樣具有重復性。該管理規程同時給出儀器分析方法在被分析物不同濃度時建議的標準回收率范圍,儀器分析方對于卵清蛋白含量檢測方法,標準回收率為66.7%-133.3%是可接受的。 對比相同一次檢測中使用一階回歸與二階回歸你和標準曲線,得到下圖:
由圖可看出,二階回歸曲線的標準品濃度X‘要比一階線性回歸得到的X要小,而對比表1中的內參數據,可以看出公司卵清蛋白含量檢測中過內參結果絕大多數均接近標準上線,即內參檢測結果偏高。如果可以將標準曲線由以及回歸改為二階回歸,即可防止出現由于內參大于標準上限導致的實驗無效。
使用卵清蛋白含量檢測試劑盒中一組標準品及內參數據,使用公司的以及回歸方法以及意大利工廠二階段回歸方法分別計算內參結果。見下表:
由表可以看出,使用二階回歸標準曲線R2可以達到0.999以上,表明該標準曲線更能代表數據的分布方式。同時內參與廠家給出的真實值更加接近。因此可以得出結論,使用二階回歸標準曲線更加準確。 |
|||||||||||||||||||||||||||||||||
|
根本原因分析 通過以上的調查分析,可以確定,本偏差發生的根本原因是:卵清蛋白含量檢測方法使用一階回歸曲線作為標注曲線,但該方法二階回歸做標準曲線更加準確,即標準曲線不充分。 |
|||||||||||||||||||||||||||||||||
|
調查組長/日期 |
|||||||||||||||||||||||||||||||||
|
QA評估組長/日期 |
|||||||||||||||||||||||||||||||||
|
生產負責人/日期 |
|||||||||||||||||||||||||||||||||
|
質量負責人/日期 |
|||||||||||||||||||||||||||||||||
5.偏差影響評估
|
偏差/事件編號 |
到期日期 |
||||||||
|
偏差/事件名稱 |
|||||||||
|
調查評估 |
|||||||||
|
調查主導部門 |
|||||||||
|
確認偏差/事件描述 充分□ 不充分□ |
|||||||||
|
是否符合國家法規或公司執照、證書要求 |
是□ 否□ |
||||||||
|
1.產品影響評估 |
有影響或潛在影響□ 無影響□ |
||||||||
|
所用潛在受影響的物料、產品等
產品影響的理由: 由于內參不合格電子計算表格回自動生成實驗室不成立的”Fail”,標志,并拒絕計算樣品含量結果,實驗員對于涉及的6批原液的卵清蛋白重新進行了檢測。因此本偏差內參不合格不會對檢測結果造成影響。 |
|||||||||
|
2.對質量管理體系的影響 |
|||||||||
|
偏差/事件對繼續生產、驗證等其他系統的影響,若無影響請說明理由: 本偏差發生在QC實驗室,不涉及生產工藝,驗證等系統無影響,對產品穩定性考察亦沒有影響,故對質量管理體系無不良影響。 |
|||||||||
|
調查組長/日期 |
|||||||||
|
QA評估組長/日期 |
|||||||||
|
生產負責人/日期 |
|||||||||
|
質量負責人/日期 |
|||||||||
偏差的兩個重要指標:偏差調査處理的及時性是偏差系統能否有效運作的關鍵因素之一,偏差報告時限以及偏差調査和處理時限是衡量偏差調査處理的及時性的兩個關鍵指標。
|
CAPA編號 |
||||
|
CAPA名稱 |
||||
|
問題或潛在風險來源 □偏差(偏差編號:_________) □事件(偏差編號:_________) □召回 □CAPA(CAPA編號______) □變更(變更編號:_________) □客戶投訴(編號_________) □產品年度回顧 □自檢 □外部審計 □工藝性能和產品質量檢測趨勢 □風險評估 □其他_____________ |
||||
|
□屬于法規承諾 □不屬于法規承若 |
||||
|
糾正預防措施CAPA |
||||
|
CAPA行動 |
計劃完成日期 |
負責人 |
||
|
更改卵清蛋白含量檢測SOP,即將標準曲線由一階改為二階回歸 |
xxxx |
xxx |
||
(來源于critical的偏差,投訴、法規承諾和檢查關鍵缺陷的CAPA必須進行有效性核查)如不需要有效性核查請說明理由 QC部門發起變更申請,變更后在下一批次的檢查測中使用變更后的方法,由于該方法在調查階段已經過充分的評估和測試,因此后續不需要進行有效性核查。 |
||||
|
CAPA開啟人/日期 |
||||
|
CAPA所用人/日期 |
||||
|
QA批準人/日期 |
||||
|
CAPA編號 |
計劃完成時間 |
|||||||||||||||||
|
CAPA名稱 |
||||||||||||||||||
|
CAPA的執行情況 □CAPA行動未全部完成 □CAPA行動已全部完成,對應的企業內部文件編號或證明性資料(附件清單,包括編號、名稱等)
□申請CAPA關閉 □CAP還需進行有效性核查
CAPA所有人/日期 |
||||||||||||||||||
|
□記錄完整、符合SOP要求 □記錄不完整、不符合SOP要求
CAPA文控專員/日期 |
||||||||||||||||||
|
□同意關閉 □不同意關閉(說明理由)
QA批準人/日期 |
||||||||||||||||||
8.CAPA的有效性核查
|
CAPA編號 |
計劃完成時間 |
|||
|
CAPA名稱 |
||||
|
有效性核查通過的要求 |
||||
|
有效性核查結果 |
□通過 □失敗 |
|||
|
□CAPA行動完成后規定的時間內是否不再發生類似的問題
□CAPA行動是否對其他工藝產生不良影響
|
||||
|
□申請CAPA關閉
□CAPA還需要采取進一步行動(如有,請給出comments)
|
||||
|
CAPA所有人/日期 |
||||
|
□記錄完整、符合SOP要求
□記錄不完整、不符合SOP要求
CAPA文控專員/日期 |
||||
|
□同意關閉
□不同意關閉(說明理由)
QA批準人/日期 |
||||
|
Critical(關鍵偏差):滿足以下任何一條即可定級為critical |
|
1.對以上是產品或用于臨床的產品安全、特性、規格、純度、效價、存在不利的影響偏差 |
|
2.質量系統重大缺陷或偶然機會識別的重大缺陷 |
|
3.可能導致病人處于污染風險 |
|
4.無菌測試失敗,包括培養基灌裝試驗 |
|
5.在確認過程中,某項程序不符合可接受標準 |
|
6.在標示的儲存條件下,產品在有效期內穩定性不合格 |
|
7.故意違背GMP(例如:偽造數據) |
|
8.病毒或細菌滅活失敗 |
|
9.產品混批和標簽錯誤(錯誤的產品名稱和規格) |
|
10.確認用于生產、檢測、和控制產品、設備、方法的驗證或者在驗證不合格 |
|
11.違背許可放行 |
|
Major: 滿足以下任何一條即可定級為major |
|
1.物料不符合披肩要求或特定的法規要求,且沒有放行的 |
|
2.不符合注冊的標準(如:中間過程控制或最終產品質量)且沒有放行的物料 |
|
3.工序微生物超行動線 |
|
4.生產期間A或B級環境結果超標 |
|
5.影響產品的注射用水或純化水結果超標 |
|
6.客戶技術性投訴的趨勢 |
|
7.中間過程參數控制不符合要求(例如:溫度、壓力、收率) |
|
8.中間過程質量不符合標準(例如:pH、蛋白質、效力、雜質) |
|
9.設別清潔失敗 |
|
10.清潔程序執行不符合要求 |
|
11.OOS |
|
12.使用過期的培養基、試劑、緩沖液等 |
|
13.GMP記錄丟失(舉例:批記錄,測試結果表) |
|
14.使用AQL檢查表對包裝產品進行檢查是,結果超標 |
|
15.成品、半成品、原輔料儲存溫度超標 |
|
16.原輔料或稱產品組分已經放行,但發現問題(如:生產時發現原輔料中有顆粒) |
|
Minor微小偏差 |
|
1.C,D級環境結果超標,且不影響產品 |
|
2.由于疏忽違背SOP,且不影響產品 |
|
3.記錄文件錯誤,且不影響產品 |
|
4.不影響工序或產品的設備故障 |
|
5.儲存溫度超溫且不影響產品 |
▲ 表1-偏差調查開啟
|
偏差/事件開啟 |
|
|
偏差或事件 |
□偏差 □事件 |
|
發現部門 |
|
|
發現事件和日期 |
|
|
發現地點 |
|
|
發生時間和日期 |
|
|
發生地點 |
|
|
通知QA的日期/時間 |
|
|
偏差/事件的客觀描述包括涉及到的產品、批次、設備編號,所違背的SOP規定等
|
|
|
立即采取的緊急措施(如有)
|
|
|
偏差/事件編號 |
到期日期 |
|
偏差/事件名稱
|
|
|
偏差調查信息 |
|
|
根本原因分析
|
|
|
調查組長/日期 |
|
|
QA評估組長/日期 |
|
|
生產負責人/日期 |
|
|
質量負責人/日期 |
|
|
偏差/事件編號 |
到期日期 |
||||||||
|
偏差/事件名稱 |
|||||||||
|
調查評估 |
|||||||||
|
調查主導部門 |
|||||||||
|
確認偏差/事件描述 充分□ 不充分□ |
|||||||||
|
是否符合國家法規或公司執照、證書要求 |
是□ 否□ |
||||||||
|
1.產品影響評估 |
有影響或潛在影響□ 無影響□ |
||||||||
|
所用潛在受影響的物料、產品等
產品影響的理由:
|
|||||||||
|
2.對質量管理體系的影響 |
|||||||||
|
偏差/事件對繼續生產、驗證等其他系統的影響,若無影響請說明理由:
|
|||||||||
|
調查組長/日期 |
|||||||||
|
QA評估組長/日期 |
|||||||||
|
生產負責人/日期 |
|||||||||
|
質量負責人/日期 |
|||||||||
|
CAPA編號 |
||||
|
CAPA名稱 |
||||
|
問題或潛在風險來源 □偏差(偏差編號:_________) □事件(偏差編號:_________) □召回 □CAPA(CAPA編號______) □變更(變更編號:_________) □客戶投訴(編號_________) □產品年度回顧 □自檢 □外部審計 □工藝性能和產品質量檢測趨勢 □風險評估 □其他_____________ |
||||
|
□屬于法規承諾 □不屬于法規承若 |
||||
|
糾正預防措施CAPA |
||||
|
CAPA行動 |
計劃完成日期 |
負責人 |
||
|
R不需要有效性核查 □需要有效性核查 (來源于critical的偏差,投訴、法規承諾和檢查關鍵缺陷的CAPA必須進行有效性核查)如不需要有效性核查請說明理由
|
||||
|
CAPA開啟人/日期 |
||||
|
CAPA所用人/日期 |
||||
|
QA批準人/日期 |
||||
|
|
|
|||||||||||||||||
|
|
||||||||||||||||||
|
||||||||||||||||||
|
|
||||||||||||||||||
|
|
||||||||||||||||||
|
CAPA編號 |
計劃完成時間 |
|||
|
CAPA名稱 |
||||
|
有效性核查通過的要求 |
||||
|
有效性核查結果 |
□通過 □失敗 |
|||
|
□CAPA行動完成后規定的時間內是否不再發生類似的問題
□CAPA行動是否對其他工藝產生不良影響
|
||||
|
□申請CAPA關閉 □CAPA還需要采取進一步行動 |
||||
|
CAPA所有人/日期 |
||||
|
□記錄完整、符合SOP要求 □記錄不完整、不符合SOP要求
CAPA文控專員/日期 |
||||
|
□同意關閉 □不同意關閉(說明理由)
QA批準人/日期 |
||||

來源:注冊圈





