依據ICH Q3A指導原則�����,藥物中的雜質可以分為有機雜質�����、無機雜質和殘留溶劑,可見殘留溶劑是藥物雜質控制的重要組成之一�����。
根據定義���,殘留溶劑是指在原料藥���、賦形劑或制劑工藝中使用或產生的揮發性有機試劑��。近年來,殘留溶劑的毒性或對環境的危害性越來越引起醫藥行業的關注����,對殘留溶劑控制的相關指導原則也越來越完善�����。
起初,1990年美國藥典僅列出了6種需要控制的殘留溶劑的限度,自1995年起我國藥典開始在附錄中收載“有機殘留量檢查”���,也只列出了7種有機溶劑,直到1997年ICH Q3C問世,收載69種在藥物中應控制的殘留溶劑�,2005年起我國藥典殘留溶劑相關通則與ICH Q3C達成一致�。從此���,殘留溶劑分為4類�����,第一類溶劑是具有致癌性或對環境有嚴重危害的溶劑���,應避免使用�����;第二類溶劑是可能引起畸變或神經毒性等不可逆毒性、或其他嚴重但可逆毒性的溶劑��,應限制使用�����;第三類溶劑是藥物中以一般量(≤0.5%)存在時認為對人體無害;第四類溶劑是尚無足夠安全性數據的溶劑���,這類溶劑的控制限度需根據文獻或數據庫提供的毒理學研究數據來確定。
對于殘留溶劑的檢測方法�,眾所周知是采用氣相色譜法�。那么氣相色譜方法開發流程是怎樣的�,有哪些經驗和技巧呢?
開發流程
第一步:全面分析待檢測藥物合成路線(包括起始物料的合成路線)中使用的有機試劑�����,除直接使用的有機溶劑外還要分析合成過程中可能會生成的有機溶劑�,這一步要盡量羅列全面,不要漏掉任何一個試劑��。例如下圖中有機試劑A至J都是需要控制的殘留溶劑����。
某原料藥合成路線簡圖
第二步:查閱待測溶劑的沸點、極性和控制限度�����,大多數常規溶劑的限度可以在中國藥典2020年版四部指導原則0861殘留溶劑測定法或ICH Q3C殘留溶劑指南中查到,如果所使用的試劑在藥典或ICH指導原則中沒有明確的限度�����,則需要查閱相關文獻或數據庫網站��,依據毒理試驗數據合理制定其控制限度��。
第三步:檢測器類型選擇,通常FID檢測器可以適用于大多數溶劑的檢測����,當待測溶劑含有強電負性時(如二氯甲烷)���,也可以選擇ECD檢測器。
第四步:溶樣溶劑選擇�����,溶樣溶劑應選擇純度高(干擾峰少)�����、對供試品溶解性好�、本身沸點略高于待測溶劑的溶劑�����,不推薦用水做溶樣溶劑��,因為水會縮短色譜柱使用壽命。最常用的溶樣溶劑包括N��,N-二甲基甲酰胺(DMF)�、二甲基亞砜(DMSO)、N-甲基吡咯烷酮(NMP)��、N,N-二甲基乙酰胺(DMA)等�����,在溶樣溶劑篩選時很難預測哪個溶劑更適宜���,需要逐個進樣后依據結果來篩選����。
第五步:進樣方式選擇��,氣相色譜進樣方式包括頂空進樣和直接進樣�,自頂空進樣問世以來��,因其對色譜柱和檢測器污染較少已成為主流,因此我們在方法開發時首選頂空進樣。
第六步:色譜柱選擇�����,根據待測溶劑的極性情況����,選擇合適的色譜柱,這也是方法開發中極其重要的一步,色譜柱填料類型及規格對待測試劑的分離起著關鍵作用。氣相色譜柱的選擇有以下四方面內容:
(1)填料類型:氣相色譜柱填料可以分為極性���、中等極性和弱極性3種。在方法開發之初,當有多種溶劑需要分離時可以首先嘗試涵蓋范圍較廣中等極性色譜柱�����,然后再分別嘗試極性和弱極性色譜柱��,這樣基本就可以實現全覆蓋��。
(2)柱長:依據范氏方程,柱長與分離度成正比。當待測有機溶劑數量較少���,樣品基質也沒有明顯干擾時,可以選擇較短的柱長��,如30 m,這樣可以縮短分析時間,提高分析效率�。當所需分析的有機溶劑數量多�、分離度不佳或者樣品基質有很多干擾峰時就需要選擇更長的柱長��,如60 m����、75m�����。例如,在某藥物殘留溶劑的分析過程中,起初采用30 m長色譜柱��,無論怎樣調整升溫程序�����,溶劑A與B之間分離度總是小于1.5�����,更換60 m色譜柱后,兩者分離度大于2.0��,滿足分離要求����。
(3)內徑:通常情況下,內徑越小��,柱效越高����,峰型越好看,但同時柱壓會相應增高�,柱容量會有所降低����。0.25 mm~0.53 mm是常用的色譜柱內徑��,內徑并不適合越細越好�����,當各種溶劑分離度都較好時可以選擇稍大的內徑,降低系統壓力,當溶劑間分離不佳需要更窄的峰型時選擇細內徑。
(4)膜厚:膜厚會影響載樣量���、分離度���、保留時間���。通常溶質保留與膜厚度成正比�,膜厚度增加可以使溶劑得到更高的保留,改善出峰較早組分的分離����,但對出峰較晚組分的分離無改善��,甚至會損失分離度。此外增加膜厚��,可以增加柱容量�,當溶劑數量多、響應偏高或進樣量較大時盡量選擇厚液膜�����,否則很容易超載���,導致峰型變差��。
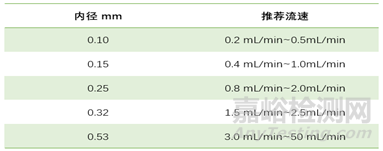
第六步:載氣流速選擇����,載氣流速的選擇相對容易�,可按最佳流速來設置,不同的內徑色譜柱對應不同的最佳載氣流速范圍�����,見下表:
通常降低流速可以提高分離度但同時會損失柱效,因此流速的調整需要在分離度改善程度和峰展寬造成的靈敏度損失之間尋找平衡�。
第七步:升溫程序設置��,像高效液相中洗脫梯度一樣���,氣相色譜中的升溫程序對分離度有重要影響�����,在方法開發之初可以先設置一個通用簡單的升溫程序�����,然后再根據各溶劑的保留情況進行調整。
經過上述的一系列操作���,我們就基本建立了一個殘留溶劑檢測的初始方法。需要注意的是��,方法開發并不是按著以上操作步驟一次性就能解決的��,往往需要在初始方法的基礎上循環往復的優化各種參數�����,以達到靈敏度、分離度���、回收率等各項要求。下面將討論當分析方法未達到目標要求時需要怎樣優化方法�?從哪些流程入手來優化��。
如何優化?
第一�����、分離度不夠
分離度是方法開發的硬性指標��,溶劑間分離度不低于1.5是不可打折扣的要求��。溶劑的保留時間及分離是溶劑沸點�����、極性�����、色譜柱填料類型以及升溫程序等各種因素的綜合作用的結果。當分離度不佳時可以考慮從以下幾方面進行優化��。
1)更換色譜柱填料類型:填料類型對溶劑的保留有重要影響���,更換色譜柱填料類型是改善分離度的最有效的方法����,例如在開發過程中如果在HP-5或DB-1701等色譜柱中總是分離度不適宜,可以考慮更換DB-624色譜柱����,可能分離度問題迎刃而解�����。
2)柱長:當在短柱中分離不好,可以更換同種填料的長柱。
3)升溫程序調整:調整的方式包括起始柱溫和升溫速率。需根據各溶劑的保留時間分布進行有針對性的調整�����,如果第一個溶劑出峰過晚或過早���,那么可以先適當提高或降低起始柱溫使第一個出峰的溶劑有一個適宜的保留時間����,然后在溶劑分布很稀疏的地方可以適當加快升溫速率,在溶劑分布很密集�、分離度不好的地方�����,可以適當降低升溫速率。
第二���、分析靈敏度不夠
一般情況下,我們所開發的方法的定量限至少應低于溶劑限度的3~5倍�,這樣更能確保限度濃度的樣品分析的準確性���。對于限度較高的溶劑如乙醇���、甲醇���、乙酸乙酯等����,這個要求很容易達到���,但當所分析的溶劑限度很低或是響應因子很低時����,往往很難達到所需的靈敏度。以苯為例來說����,其限度是0.0002%(2 ppm)�����,定量限需要達到的濃度應為0.00004%~0.00007%(0.4ppm~0.7ppm)之間��,這是一個挑戰�。這時想要改善靈敏度可以從以下幾方面入手:
1)提高供試品的濃度:溶劑的限度是相對于供試品濃度計算的���,提高供試品濃度也就相當于提高了溶劑限度的絕對濃度���,響應也就相應提高了��。
2)調整分流比:一般情況下����,為保護色譜柱和檢測器免受污染�����,分流比設置為10:1�����,也就是只有10%的樣品進入色譜柱和檢測器,當溶劑限度很低時��,可以把分流比設置成5:1或2:1等����,使流入色譜柱和檢測器的樣品比例更高,響應自然也提高了�。
3)提高頂空平衡溫度:頂空平衡溫度越高��,達到氣液平衡時溶劑的濃度越高,我們在實際的方法開發中��,發現當頂空溫度由90℃提高到110 ℃時�,苯的峰面積提高了約1.6倍�。
4)改為直接進樣:如果頂空進樣無法滿足要求,可以改為直接進樣,靈敏度會大大提高,但是直接進樣對色譜柱污染嚴重����,應慎重選擇��。
第三、回收率偏低或偏高
造成回收率不符合要求的原因通常是基質效應����,包括基質減弱效應和基質增強效應�����,造成的結果是回收率偏低和回收率偏高�����。
(1)回收率偏低
造成回收率低的原因包括供試品對待測溶劑有吸附作用、供試品與待測溶劑發生反應和待測溶劑沸點過低�,揮發損失等��,相信大家在實際方法開發中遇到最多的基質減弱效應,也就是回收率偏低�。
(2)回收率偏高
除了基質減弱效應���,還有基質增強效應��,結果是回收率偏高,其原因是供試品占據了色譜系統中部分活性位點���,減少了色譜系統與待測溶劑的反應,造成待測溶劑響應偏高�����。
解決基質增強效應的方法包括1)在靈敏度允許的條件下適當降低供試品溶度���;2)加入一定量的分析保護劑�����,保護劑會與色譜系統中的活性位點發生反應,達到保護殘留溶劑的作用。例如:某殘留溶劑回收率偏高�,約為120%����,當向溶液中添加一定量醇類保護劑后����,待測溶劑的回收率為103%左右,符合要求;3)當以上方法不能解決時,可以采用終極方法,即采用標準加入法測定回收率�����,這樣可以完全消除對照溶液與供試品溶液基質的差別�。
要而論之,分析條件優化的目的是采用最簡便的操作、在最短的時間內達到符合要求的分離度���、靈敏度和準確度。如果在初始條件下難分離對的分離度就符合要求�,那么我們可以通過調整升溫程序��、流速等手段縮短分析時間,否則�,我們應設法提高分離度����,當然這個過程可能會損失一定的靈敏度和分析時間���,我們所要做的是尋找其中的平衡點����。
以上是我對于殘留溶劑方法開發的一些經驗��,希望能對大家有幫助�����。
參考文獻:
[1]中國藥典2020年版四部通則0861
[2] 毛婭,頂空-氣相色譜法同時測定藥品包裝材料中30種溶劑殘留量���,中國藥師,2022年第25卷第12期�,2280~2285����。
[3] 張志杰,氣相色譜分析方法的開發����,遼寧化工����,2006年5月第35卷�����,第5期��,302~305。
[4] Colin F. Poole����,Matrix-induced response enhancement in pesticide residue analysis by gas chromatography��,Journal of Chromatography A, 1158 (2007) 241–250。
[5] Claudia Witschi���,Residual solvents in pharmaceutical products: acceptable limits, influences on physicochemical properties, analytical methods and documented values�,European Journal of Pharmaceutics and Biopharmaceutics,Volume 43, Issue 3, June 1997, Pages 215-242
[6] KrakÛw, Poland, Analytical methods for residual solvents determination in pharmaceutical products,Acta Poloniae Pharmaceutical Drug Research, Vol. 67 No. 1,13~26, 2010.