剛剛,國家藥監局發布《丹七片中異性有機物檢查項補充檢驗方法》《腦立清丸(膠囊、片)中水麥冬酸檢查項補充檢驗方法》《檀香清肺二十味丸中松香酸檢查項補充檢驗方法》《小柴胡顆粒中黃芩提取物檢查項補充檢驗方法》,內容如下:
丹七片中異性有機物檢查項補充檢驗方法
(BJY 202301)
【檢查】異性有機物 本品為部分浸膏片,除三七顯微特征外,不得檢出其他植物組織。
取本品2片,除去包衣,研細,稱取細粉0.1g,加水10ml使溶解,離心,棄去上清液,沉淀加水2ml使混懸,吸取混懸液1滴,置于載玻片上,加水合氯醛1滴,加熱透化,置顯微鏡100倍下,按下圖選取9個檢查點檢視,視野中除三七的植物組織特征外,不得檢出其它植物組織。
如僅有1個檢查點視野中檢出除三七顯微特征外的其他植物組織,應依法制片復試,復試不得檢出。
蓋玻片上檢查點示意圖
起草單位:柳州市質量檢驗檢測研究中心(柳州市食品藥品檢驗檢測中心)
復核單位:湖南省藥品檢驗檢測研究院
北京市藥品檢驗研究院
腦立清丸(膠囊、片)中水麥冬酸檢查項補充檢驗方法
(BJY 202302)
【檢查】水麥冬酸 照高效液相色譜法(中國藥典 2020 年版通則 0512)和質譜法(中國藥典 2020 年版通則 0431)測定。
色譜、質譜條件與系統適用性實驗 以十八烷基硅烷鍵合硅膠為填充劑(色譜柱內徑2.1mm);以乙腈為流動相A,以0.1%甲酸溶液為流動相B,按下表中的規定進行梯度洗脫,流速為每分鐘0.2ml;采用質譜檢測器,電噴霧負離子模式(ESI-),進行多反應監測(MRM),選擇質荷比m/z187.0→143.0和m/z187.0→99.0作為檢測離子對。取對照品溶液,進樣2µl,按上述離子對測定的MRM色譜峰的信噪比均應大于10:1。
|
時間(分鐘)
|
流動相A(%)
|
流動相B(%)
|
|
0~10
|
1→5
|
99→95
|
對照品溶液的制備(臨用新制) 取水麥冬酸對照品適量,精密稱定,加乙腈-0.1%甲酸溶液(1:99)制成每1ml含0.25μg 的溶液,作為對照品溶液。
供試品溶液的制備 取腦立清丸、片、膠囊內容物,研細,取約8.0g(相當于清半夏1g),精密稱定,置具塞錐形瓶中,精密加水50ml,稱定重量,超聲處理(功率500W,頻率40kHz)45分鐘,放冷,再稱定重量,用水補足減失的重量,搖勻,離心(轉速為每分鐘8000轉)5分鐘,取上清液,濾過,精密量取續濾液25ml至離心管中,加甲酸0.2ml,搖勻,加入乙酸乙酯20ml,搖勻,離心(轉速為每分鐘5000轉)5分鐘,分取乙酸乙酯液,自“加入乙酸乙酯20ml”起,再同法操作3次,合并乙酸乙酯液,減壓回收溶劑至干,殘渣加乙腈-0.1%甲酸溶液(1:99)使溶解,并定容至2ml,用微孔濾膜(0.22μm)濾過,取續濾液,即得。
測定法 分別精密吸取對照品溶液與供試品溶液各2μl,注入液相色譜-質譜聯用儀,測定,即得。
(1)供試品的提取離子流色譜中,未同時出現與對照品溶液色譜相應的色譜峰,視為未檢出;(2)供試品的提取離子流色譜中,同時出現與對照品溶液色譜相應的色譜峰,且供試品色譜中 m/z 187.0→143.0 的色譜峰面積值不大于對照品溶液中相應的峰面積值,視為未檢出;(3)供試品的提取離子流色譜中,同時出現與對照品色譜相應的色譜峰,且供試品色譜中 m/z 187.0→143.0的色譜峰面積值大于對照品溶液中相應的峰面積值,視為檢出。
結果判定 供試品的提取離子流色譜中,應不得檢出與對照品溶液色譜相應的色譜峰。
起草單位:山東省食品藥品檢驗研究院
復核單位:河北省藥品醫療器械檢驗研究院
四川省藥品檢驗研究院(四川省醫療器械檢測中心)
檀香清肺二十味丸中松香酸檢查項補充檢驗方法
(BJY 202303)
【檢查】松香酸 照高效液相色譜法(中國藥典2020年版通則0512)測定。
色譜條件與系統適用性試驗 以十八烷基硅烷鍵合硅膠為填充劑;以乙腈-0.1%甲酸(74:26)為流動相;檢測波長為241 nm。理論板數按松香酸峰計算應不低于3000。
對照溶液的制備(臨用新制) 取松香酸對照品適量,精密稱定,加乙醇制成每1ml含2µg的溶液,作為對照品溶液。另取11-羰基-β-乙酰乳香酸對照品適量,精密稱定,加乙醇制成每1ml含2µg的溶液,作為參照溶液。
供試品溶液的制備 取本品,研細,取0.2g,精密稱定,精密加入乙醇20ml,稱定重量,超聲處理20分鐘,放冷,再稱定重量,用乙醇補足減失的重量,搖勻,濾過,取續濾液,即得。
測定法 分別精密吸取供試品溶液、對照品溶液與參照溶液各10µl,注入液相色譜儀,記錄色譜圖。
結果判定 供試品色譜中,在與松香酸對照品溶液色譜峰保留時間相應的位置上不得出現相同的色譜峰。若出現保留時間相同的色譜峰,采用二極管陣列檢測器比較相應色譜峰的紫外-可見吸收光譜,吸收光譜應不同(松香酸對照品色譜峰在241nm顯示最大吸收);若吸收光譜相同,且該色譜峰的峰面積大于11-羰基-β-乙酰乳香酸參照溶液色譜峰的峰面積值,則視為陽性檢出。
備注:必要時,可采用高效液相色譜-質譜聯用方法驗證。
起草單位:連云港市食品藥品檢驗檢測中心
復核單位:海南省藥品檢驗所
云南省食品藥品監督檢驗研究院
小柴胡顆粒中黃芩提取物檢查項補充檢驗方法
(BJY 202304)
【檢查】黃芩提取物 照高效液相色譜法(中國藥典2020年版通則0512)測定。
色譜條件與系統適用性試驗 以十八烷基硅烷鍵合硅膠為填充劑(建議色譜柱的內徑為4.6mm,粒徑為2.7μm);以甲醇為流動相A,0.5%甲酸為流動相B,按下表中的規定進行梯度洗脫;流速為每分鐘0.6ml;檢測波長為270nm。理論板數按黃芩苷峰計算應不低于5000。
|
時間(分鐘)
|
流動相A(%)
|
流動相B(%)
|
|
0~10
|
5→25
|
95→75
|
|
10~40
|
25→55
|
75→45
|
|
40~55
|
55→80
|
45→20
|
參照物溶液的制備 取黃芩對照藥材0.1g,加水煎煮1.5小時,濾過,濾液濃縮至近干,加入50%乙醇溶液25ml,密塞,超聲處理(功率350W,頻率37kHz)45分鐘,取出,放冷,搖勻,濾過,濾液用0.22μm微孔濾膜濾過,作為對照藥材參照物溶液。另取黃芩苷對照品和漢黃芩苷對照品適量,加甲醇制成每1ml各含60µg的混合對照品溶液,搖勻,用0.22μm微孔濾膜濾過,作為對照品參照物溶液。
供試品溶液的制備 取本品,混勻,研細,取約1g﹝規格(1)﹞、0.4g﹝規格(3)﹞、0.3g﹝規格(2)、規格(4)﹞或0.25g﹝規格(5)﹞(均相當于含黃芩生藥量0.056g),精密稱定,置具塞錐形瓶中,精密加入50%乙醇溶液25ml,密塞,稱定重量,超聲處理(功率350W,頻率37kHz)45分鐘,取出,放冷,再稱定重量,用50%乙醇溶液補足減失的重量,搖勻,濾過,濾液用0.22μm微孔濾膜濾過,即得。
測定法 分別吸取參照物溶液與供試品溶液各5μl,注入超高效液相色譜儀,測定,即得。
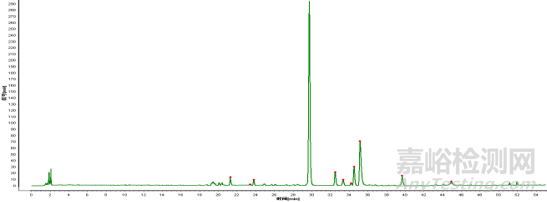
結果判定 供試品色譜中應呈現與對照藥材參照物中5個主要特征峰保留時間相對應的色譜峰,其中峰1與峰4應與對照品參照物峰保留時間一致,且峰4與峰1的峰面積比值應不低于0.10。
對照特征圖譜
5個特征峰中 峰1:黃芩苷;峰4:漢黃芩苷;峰5:黃芩素
注:規格(1)每袋裝10g;(2)每袋裝5g(無蔗糖);(3)每袋裝4g(無蔗糖);(4)每袋裝3g(無蔗糖);(5)每袋裝2.5g(無蔗糖)。
起草單位:廣東省藥品檢驗所
復核單位:湖南省藥品檢驗檢測研究院