您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2022-10-01 07:45
開展強制降解試驗是獲得藥物可能的降解途徑和降解產物信息的重要途徑,有助于更全面了解產品的雜質譜。對于分析方法的開發和質量標準的建立具有重要意義,是雜質檢測方法建立、優化和驗證的重要研究手段。可為藥品的處方、工藝、包裝、貯藏條件的確定提供有益支持。
1 關于強制降解的程度
一般5%~20% 的降解較為合適,需避免二次降解。對于難以降解的、非常穩定的化合物,應提供合理解釋和判斷依據。《化學藥物雜質研究技術指導原則》中也指出,破壞試驗的程度暫無統一要求,一般以強力破壞后主成分的含量仍占絕大部分為宜。此時已產生了一定量的降解產物,與樣品長期放置的降解情況相似,考察此情況下的分離度更具有實際意義。要達到這種破壞程度,需要在研究過程中進行摸索,先通過文獻調研、理論分析或預試驗了解樣品對光、熱、濕、酸、堿、氧化條件的基本穩定情況,然后優化確定破壞性試驗條件(如光照強度、酸堿濃度、破壞的時間、溫度等),以得到能充分反映降解產物與主成分分離的結果和圖譜。
關于破環試驗的降解量。通常認為降解量應在5-20%之間。對于含量限度為標示量的90%-110%的小分子藥物,即使在文獻中有更廣泛的推薦范圍作為參考(例如,10-30%),通常允許10%的降解量。對于非常穩定性的藥物,如何破壞都不降解,還遇到這樣的發補:請繼續加強對制劑降解途徑和降解雜質的研究。怎么辦。
2 關于強制降解的時間
(1)加強對制劑降解途徑的研究
藥物被破壞達到一定的時間后依然沒有降解,無需再研究。多少時間,10天,見下表(來源WHO)。

(2)加強對降解雜質的研究
從數個項目發補內容可知,破壞試驗產生的降解雜質是否研究,CDE也不統一。即使不統一,研究階段還是要研究(沒有研究透徹也不影響申報,申報以后再繼續研究,發補時提交資料)。加強對降解雜質的研究,言外之意是鑒定雜質結構。有一個簡化的做法,參考FDA QBD模板(見下表),雜質F是未知雜質(僅采用RRT定位),限度超過鑒定限,也不鑒定結構,為什么。通過HPLC保留時間、UV(DAD),以及MS(分子量),確定雜質F是自制品與RLD的共有雜質,,沒有安全性風險。
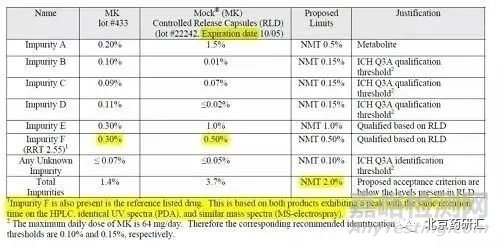
超過鑒定限的未知雜質只需用HPLC保留時間、UV(DAD),以及分子量(一級和二級質譜圖),證明與RLD為共有雜質,則無需鑒定結構。
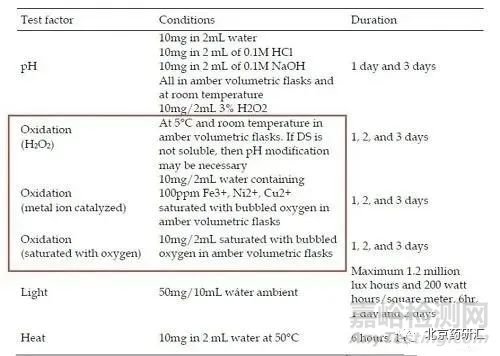
(3)氧化破壞的破壞方式
雙氧水氧化(常見)、金屬離子催化(在Fe3+, Ni2+, Cu2+的溶劑,通氧氣使溶液中的氧處于飽和狀態)、飽和氧環境(通氧氣使溶液中的氧處于飽和狀態)
(4)酸/堿水解
水解反應是物質與水發生的導致物質發生分解的反應,即物質與水中的氫離子或者氫氧根離子發生反應。大多數有機化合物的水解,僅用水是很難順利進行的,一般需在堿性或酸性條件下進行,這是由于水解反應的發生需克服水解基團的水解活化能(activation energy,Ea),即需要考慮水解基團所處的電子和空間位阻效應。如藥物分子中含有的羧酸酯、羧酸酰胺及磷酸酰胺等基團通常水解活化能較低(Ea 通常< 20 kcal·mol-1 ),是比較容易水解的位點; 如醚及磺酰胺基團,一般水解活化能較高(Ea 通常> 30 kcal·mol-1 ),通常不易發生水解。
酸/堿水解考察條件的設置主要考慮因素包括:酸/堿溶液的濃度(或pH 值)、考察的溫度與時間,具體考察條件需根據藥品特點,特別是分析藥物結構中含有的水解基團及其所處的電子和空間位阻環境。如對于含有羧酸酯的藥物,其可能對堿水解十分敏感,就可使用較低濃度的氫氧化鈉溶液,在室溫條件下進行考察即可。而同樣對于含有羧酸酯的藥物,如果所處空間環境位阻較大,如叔丁基酯,可能水解條件需適當加強。
另外,一些在水溶液中溶解度不好的親脂性藥物,需注意添加適當的有機溶劑進行增溶,不應一味地增大破壞強度,造成次級降解。常用水解考察條件包括: 0.1~1 mol·L-1 的鹽酸或氫氧化鈉溶液,在室溫或加熱條件下進行考察,如60 ℃ /2 d。
(5) 光照降解
光照強制降解試驗的條件設置在ICH Q1B中有較明確規定,可分別在樣品均質化或溶液狀態下進行考察,一般要求總照度不低于1. 2 × 106 Lux·h(冷白光燈)或近紫外能量不低于200 w·h·m-2(紫外燈),如254 或365 nm 光源照射24 h。
需注意將光照發熱對受試樣品的影響降到最低,還應考慮樣品的物理性質,并應采取措施如冷藏和/或置密閉容器中,以確保物理狀態變化(如升華、蒸發、熔化)所造成的影響最小。
(6) 高溫降解
對于熱降解,一般遵循阿倫尼烏斯(Arrhenius)方程,即隨溫度升高降解速率加快。高溫降解試驗即運用這一原理,通過設置較高的考察溫度在較短的時間內獲得藥品的降解信息。具體考察溫度和時間需根據藥品特點,在前期預試驗的基礎上靈活確定,常見如80 ℃ /10 d,130 ℃ /8 h。也常結合濕度進行設置,如對于原料藥或固體制劑通常采用相對濕度75% 或更高(如80 ℃ /92. 5% RH等),而對于液體或半固體制劑可能需考慮采用干熱條件(低濕度),如相對濕度25%或更低。受試樣品可分別在固體和溶液狀態下進行考察,需注意涵蓋生產過程中最差條件的考察,如含主藥的料液在噴霧干燥過程中溫度升高,又如半固體制劑水相或油相溶解主藥過程中可能需升高溫度。對于破壞程度過高或過低的情況,可能還需進一步結合更接近于實際高溫情況的影響因素試驗(如60 ℃ /75%RH/1 個月)的考察結果,來綜合判斷是否藥物本身對熱特別穩定或特別敏感。
(7)物料平衡
下表是兩種計算方法有不同的角度,但不僅限于兩種,可自行選擇適合的計算
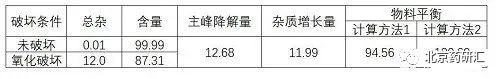
計算方法1:雜質增長量÷主峰降解量×100
計算方法2:100-主峰降解量+雜質增長量
如果存在物料不守恒(含量下降5%,總雜小于1%),通常有兩個假設:
假設1:溶液提取不充分。
思路:通過不同溶劑提取(酸、堿、甲醇、乙腈、DMSO等)、超聲的時間和溫度提取。
假設2:降解物未檢出。
思路:降解物高水溶液-無保留,采用親水色譜體系;高脂溶性-未洗脫,采用高比例有機相梯度洗脫;無紫外吸收,更換檢測器如ELSD、CAD等。
備注:各已知雜質與主峰的分離度滿足要求的情況下,含量測定方法驗證無需考察破壞試驗。

來源:Internet


