您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2022-08-26 11:10
一、前言
仿制藥是指與原研藥具有相同活性成分、劑型、給藥途徑和治療效果的替代原料藥及其制劑,仿制藥應與原研藥或參比制劑治療等效。FDA 在《橙皮書》中規定,治療等效的仿制藥要滿足以下5條總體原則:
① 與原研產品具有一樣的安全性和有效性。
② 藥學等效。
③ 生物等效(且藥品本身不存在生物等效實驗方法學的問題,或雖有問題但不影響生物等效的結果判定)。
④ 適當的說明書。
⑤ 按照 cGMP 的要求組織生產。
2016年3月5日,CFDA轉發了國務院辦公廳發布的《關于開展仿制藥質量和療效一致性評價的意見》,意味著一致性評價的大幕已正式拉開。隨著藥品審評審批制度的改革及流程的優化,仿制藥一致性評價申請觸發的研制生產現場檢查工作也有了大幅地變化。
二、現場檢查工作程序
在《總局關于調整藥品注冊受理工作的公告》(2017 年第134 號)發布以前,仿制藥注冊申請由各省局受理,并由省局進行藥學研制現場和生產現場檢查后轉入藥品審評中心進行審評(如圖1)。自2017 年12 月1 日集中受理實施后,根據藥品技術審評中的需求,由食品藥品審核查驗中心(以下簡稱“核查中心”)統一組織全國藥品注冊檢查資源實施現場核查(如圖2)。
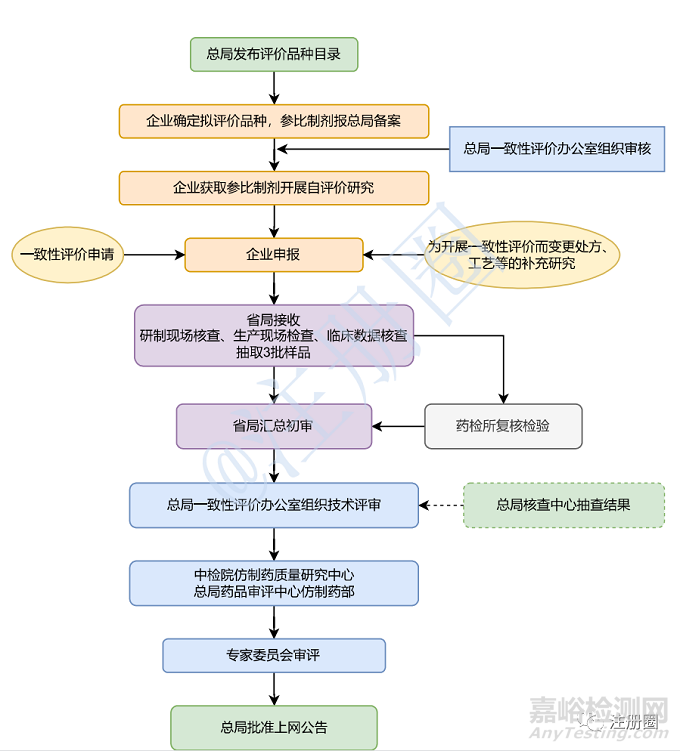
圖1 仿制藥一致性評價工作程序(逢審必查)
2016年5月26日,《總局關于發布仿制藥質量和療效一致性評價工作程序的公告》(2016年第105號)發布,規定國產仿制藥由省級食品藥品監督管理部門負責本行政區域內一致性評價資料的接收和相關補充申請資料的受理,組織研制現場核查和生產現場檢查,現場抽取連續生產的三批樣品送指定的藥品檢驗機構進行復核檢驗。完成上述工作后,由省級食品藥品監督管理部門匯總報送一致性評價辦公室。
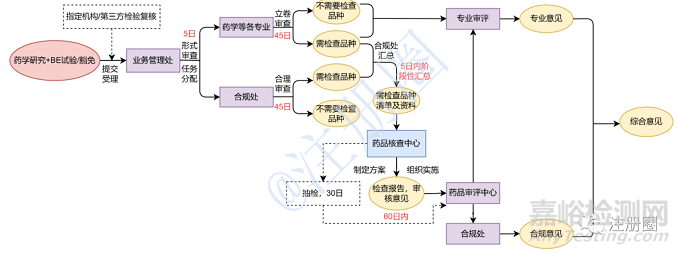
圖2 仿制藥一致性評價工作程序(基于審評、風險需要)
2017 年8 月25 日,《總局關于仿制藥質量和療效一致性評價工作有關事項的公告》(2017年第100 號)發布,要求一致性評價申請受理后,藥品審評中心于45日內對申報資料進行立卷審查,并根據立卷審查和風險評估情況提出有因檢查和抽檢的需求;藥品審評中心設立合規辦公室,協調有因檢查、抽檢以及審評等相關環節;核查中心組織對研制現場、生產現場或臨床試驗數據的有因檢查或抽樣。
由此可見,仿制藥一致性評價現場檢查工作由原先的“逢審必查,串聯程序”調整為“基于審評需要”,再到2020年版的《藥品注冊管理辦法》規定的“基于風險啟動,并聯開展”,現場檢查工作程序日漸科學合理。
三、現場檢查工作流程
2022年1月1日起施行的《藥品注冊核查工作程序(試行)》規定,藥品審評中心基于技術審評需要,啟動藥品注冊現場核查,申請人在接收到藥品審評中心發出的《注冊現場核查通知》后,需登錄核查中心藥品注冊申請人之窗,填報相關資料,并做好迎檢準備,配合完成現場檢查工作。
3.1、現場檢查的確認(填報)
法規要求:《關于化藥仿制藥質量和療效一致性評價注冊申請開展藥學研制和生產現場檢查有關事項的通告》(2020年第4號)
線上填報:《仿制藥質量和療效一致性評價品種研制生產情況申請表》(以下簡稱《申請表》)
其他附件:需上傳藥品GMP證書、藥品生產許可證復印件、《現場主文件》、《現場檢查準備情況》(模板見“2017年第77號”)
紙質遞交:打印《申請表》及其附件3份,加蓋申請人公章
3.2、迎檢準備
法規要求:《企業指南:仿制藥質量和療效一致性評價藥學研制現場檢查要求》、《企業指南:仿制藥質量和療效一致性評價生產現場檢查要求》
表1 仿制藥一致性評價研制生產現場檢查準備清單
|
迎檢準備 |
研制現場核查 |
生產現場檢查 |
|
首次會議企業匯報資料 (PPT) |
1. 藥品研制/生產基本情況 (如屬委托,應說明被委托研究單位基本情況) |
|
|
2. 一致性評價工作所涉及的所有藥品生產批次(含BE批、工藝驗證批)生產的地址、生產線、批量、生產時間、地點、使用量和剩余量等 |
||
|
3. 一致性評價所涉及的所有生產批次(含BE批、工藝驗證批)所用處方、生產工藝、原輔料、包裝材料來源及標準、生產線(設備設施)、產品質量標準(含中間控制標準)等是否與已上市/擬上市商業化生產規模的批次一致 |
||
|
4. 參比制劑的來源、采購和使用情況 |
||
|
5. 藥品和參比制劑體外研究的對比情況,包括時間、批號和結果 |
||
|
6. 藥品關鍵質量屬性(含穩定性)變化情況 |
||
|
7. 質量體系風險情況:包括品種風險,工藝風險及設備風險等 |
||
|
— |
8. 質量體系運行情況:組織機構和質量管理簡介,藥品GMP執行情況,近3年藥品GMP檢查缺陷整改情況 |
|
|
— |
9. 商業批生產線的設備、設施、生產規模情況,與其他品種共線生產情況及風險評估的結果 |
|
|
— |
10. 檢查品種動態生產安排情況 |
|
|
迎檢準備 |
研制現場核查 |
生產現場檢查 |
|
文件資料 (原始文件或原始文件的真實拷貝) |
注冊申報資料(全套) |
|
|
委托研究協議和質量協議(如有) |
委托生產協議和質量協議(如有) |
|
|
參比制劑的來源及證明 包括: 1. 購買發票、贈送證明等 2. 參比制劑的包裝標簽、說明書、剩余樣品等 3. 參比制劑的接收、發放、使用記錄或憑證 |
||
|
關鍵儀器設備 包括名稱、型號、內部編號及所在位置信息等 |
||
|
溶出度儀的驗證資料 |
||
|
一致性評價工作所涉及的產品的體外評價資料 體外研究報告內容至少包括研究時間,研究批次,研究項目,結果對比,結論等 |
||
|
一致性評價工作所涉及產品的剩余樣品情況(不應銷毀) |
||
|
藥品相關研究記錄 包括: 1. 處方工藝研究原始記錄(如有) 2. 樣品試制相關原始記錄 3. 質量研究相關原始記錄 4. 體外評價及穩定性研究的相關原始記錄 5. 儀器設備使用記錄 6. 紙質圖譜和/或電子圖譜 |
藥品生產相關規程 包括: 1. 生產工藝規程(若處方工藝未變更,還需提供根據最近一次批準的注冊申請制定的生產工藝規程) 2. 標準操作規程(產品相關生產操作規程、設備操作規程、原輔料取樣檢驗操作規程) 3. 原批準的質量標準和申報的質量標準 4. 空白批生產記錄(批生產主記錄) |
|
|
藥品檢驗方法確認或驗證資料 |
工藝驗證方案和報告,以及設備確認、清潔驗證情況 |
|
|
穩定性試驗方案及報告 |
— |
|
|
一致性評價工作所涉及的所有生產批次(含BE批、工藝驗證批等) 1.相關記錄,包括:批生產記錄、批檢驗記錄、穩定性試驗記錄、儀器設備使用記錄、紙質圖譜和/或電子圖譜 2. 原輔料、內包材供應商檔案 3. 物料臺賬及相關單據 |
||
|
接受現場檢查品種: 1. 近3年的年度質量回顧報告 2. 生產線近3年接受境內外檢查機構檢查情況及整改資料 |
||
|
注意:申請人應盡可能將不同場地的紙質文件(包括原始記錄)集中,以便查閱 |
||
|
迎檢準備 |
研制現場核查 |
生產現場檢查 |
|
人員要求 |
藥品研制總負責人、關鍵試驗項目研究負責人、研究人員、樣品試制、樣品檢驗人員;研究機構質量保證負責人和相關人員 |
生產過程所涉及的生產、檢驗、質量保證相關人員、企業質量負責人 |
|
藥品注冊負責人 |
||
|
熟悉檢測設備各項功能并具備系統管理權限的人員 |
||
|
原材料、樣品、參比制劑各類原始記錄、檔案資料、票證憑據等的保管人員、財務人員 |
||
3.3、配合檢查及后續工作
申請人收到現場核查通知后,如涉及動態生產現場檢查的,應及時排產;收集備查的紙質文件,提前對文件資料的內容熟知,以便能及時找出核查老師需要的文件資料;保證檢查中相關人員到位,并做好核查老師的接待工作。現場檢查結束后,申請人應按通知書要求送檢;跟蹤檢驗機構結果,及時溝通。
四、現場核查要點
依據《總局關于發布仿制藥質量和療效一致性評價研制現場核查指導原則等4個指導原則的通告》(2017年第77號),仿制藥一致性評價研制生產現場檢查的基本要求是,真實性、一致性、數據可靠性及合規性。
研制現場核查的目的是對藥學研究情況(包括處方與工藝研究、樣品試制、體外評價等)進行實地確證,對原始記錄進行審查,確認申報資料真實性、一致性和數據可靠性,以及研制過程合規性的過程。


圖3 仿制藥一致性評價研制現場核查要點
生產現場檢查的目的是對申報品種的生產條件和能力及其動態生產過程進行檢查,確認相關生產和質量控制活動與申報的處方、生產工藝、生產條件、質量標準的一致性,以及藥品生產是否符合《藥品生產質量管理規范》要求。


圖4 仿制藥一致性評價生產現場核查要點
五、現場檢查判定原則
表2 仿制藥一致性評價研制生產現場檢查結果判定
|
核查結論 |
判定原則 |
|
|
研制現場核查 |
生產現場檢查 |
|
|
“通過” |
未發現真實性問題、且與申報資料一致的 |
|
|
“不通過” |
發現真實性問題 |
|
|
存在與申報資料不一致 |
||
|
存在嚴重的數據可靠性問題的 |
||
|
缺少原始記錄導致無法溯源 |
||
|
不配合檢查,導致無法繼續進行現場檢查 |
||
|
— |
生產過程嚴重不符合《藥品生產質量管理規范》 |
|
參考來源
[1] 淺談仿制藥質量一致性評價 https://zhuanlan.zhihu.com/p/129737945
[2] 仿制藥一致性評價現場檢查工作進展及新法規下藥品注冊核查的思考.《中國食品藥品監管雜志》
https://ypjd.cbpt.cnki.net/WKE3/WebPublication/paperDigest.aspx?paperID=856671dd-f0e1-4c28-86d4-f3d859706b0c

來源:注冊圈


