您當(dāng)前的位置:檢測(cè)資訊 > 科研開發(fā)
嘉峪檢測(cè)網(wǎng) 2022-05-24 11:02
據(jù)Simon Smith[1]的統(tǒng)計(jì),截至2021年5月,全球共有43家制藥公司明確在藥物設(shè)計(jì)中使用人工智能,全球十大藥企均榜上有名(如圖1所示),足以見得人工智能在藥物設(shè)計(jì)中的重要性。
人工智能(AI),特別是其中的深度學(xué)習(xí),為創(chuàng)新藥物的設(shè)計(jì)提供了機(jī)會(huì)。近幾年出現(xiàn)了各種機(jī)器學(xué)習(xí)方法,其中一些被認(rèn)為是特定領(lǐng)域的人工智能實(shí)例,并且許多人工智能已成功地用于藥物設(shè)計(jì)。
本文先概述了4個(gè)有代表性的人工智能在藥物設(shè)計(jì)中的應(yīng)用,包括三種藥物設(shè)計(jì)方法以及藥物構(gòu)效關(guān)系解釋,接著討論了人工智能中的藥物設(shè)計(jì)趨勢(shì),希望可以為各位讀者提供藥物設(shè)計(jì)潛在未來(lái)方向的參考。
1針對(duì)靶標(biāo)蛋白的新藥設(shè)計(jì)
近年來(lái),基于深度學(xué)習(xí)的方法成為新藥設(shè)計(jì)的一種很有前途的工具。這些方法大多是基于配體的,其中初始的靶標(biāo)特定的配體數(shù)據(jù)集,對(duì)于設(shè)計(jì)具有優(yōu)化性質(zhì)的有效分子是必要的。盡管已經(jīng)有人嘗試開發(fā)另一種方法來(lái)設(shè)計(jì)靶標(biāo)特定的配體數(shù)據(jù)集,但在設(shè)計(jì)針對(duì)新靶標(biāo)蛋白的分子時(shí),此類數(shù)據(jù)集的可用性仍然是一個(gè)挑戰(zhàn)。
Sowmya, Navneet[4]等人提出了針對(duì)靶標(biāo)蛋白活性部位的深度學(xué)習(xí)方法。首先,使用圖形注意力模型來(lái)學(xué)習(xí)實(shí)驗(yàn)上已知的形成蛋白質(zhì)−配體復(fù)合體的蛋白質(zhì)活性部位,了解其氨基酸的結(jié)構(gòu)和特征。接下來(lái),將學(xué)習(xí)到的活性中心特征與預(yù)先訓(xùn)練的生成模型一起用于有條件地生成新分子。然后在強(qiáng)化學(xué)習(xí)框架中使用生物活性預(yù)測(cè)模型,來(lái)優(yōu)化條件生成模型(如圖2所示)。

圖2. 基于蛋白質(zhì)結(jié)構(gòu)的藥物設(shè)計(jì)流程,來(lái)源:參考文獻(xiàn)4
最后,用兩種蛋白質(zhì)Janus kinase2(JAK2)和多巴胺受體D2(DRD2) 驗(yàn)證了方法,在這兩種蛋白質(zhì)中,產(chǎn)生了與已知抑制劑類似的分子。圖形注意力模型可以識(shí)別可能的關(guān)鍵活性部位殘基,從而影響條件分子生成器設(shè)計(jì)具有與已知抑制劑相似的藥理特征的新分子。
2基于配對(duì)多目標(biāo)優(yōu)化的新藥設(shè)計(jì)
多目標(biāo)優(yōu)化解決了藥物的自動(dòng)化從頭設(shè)計(jì)問(wèn)題,這是人類創(chuàng)造過(guò)程的結(jié)果。Alberga, Gambacorta[5]等人設(shè)計(jì)了一種新的配對(duì)多目標(biāo)方法,該方法在基于遞歸神經(jīng)網(wǎng)絡(luò)的自適應(yīng)生成算法中實(shí)現(xiàn),用于自動(dòng)從頭設(shè)計(jì)新藥物分子,其總體特征通過(guò)在相關(guān)物理化學(xué)性質(zhì) (MW、logP、HBA、HBD) 和偏向特定生物靶標(biāo)的約束之間找到最佳權(quán)衡來(lái)優(yōu)化。
他們進(jìn)行了針對(duì)SARS-CoV-2主要蛋白酶、乙酰膽堿酯酶、神經(jīng)氨酸苷酶的藥物分子的從頭設(shè)計(jì)。采用幾種質(zhì)量指標(biāo)(包括有效性、唯一性、內(nèi)部多樣性、過(guò)濾值、綜合可達(dá)性)來(lái)評(píng)價(jià)藥物的相似性、化學(xué)可行性、多樣性和有效性。最后進(jìn)行了分子對(duì)接,以進(jìn)一步評(píng)估新生成的抑制劑對(duì)相應(yīng)所需生物靶標(biāo)的有效性情況(如圖3所示)。
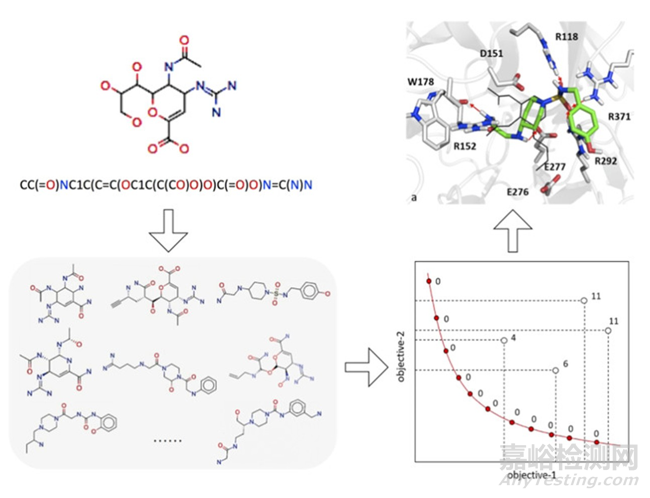
圖3. 多目標(biāo)優(yōu)化方法流程圖,來(lái)源:參考文獻(xiàn)5
結(jié)果表明,多目標(biāo)優(yōu)化能夠捕捉連接化學(xué)和生物方面的潛在聯(lián)系,從而為可定制的設(shè)計(jì)策略提供易于使用的選擇,這對(duì)線索生成和線索優(yōu)化都特別有效。
3深度學(xué)習(xí)與水藥效團(tuán)模型
相結(jié)合的藥物設(shè)計(jì)
在確定了一個(gè)靶點(diǎn)后,尋找苗頭化合物是基于結(jié)構(gòu)的藥物設(shè)計(jì)的重要第一步,常用的方法有虛擬篩選、組合化學(xué)、高通量篩選、DNA編碼化合物庫(kù)篩選等。Minsup, Kichul[6]等人設(shè)計(jì)了一種靶向特定的藥物設(shè)計(jì)方法,該方法利用了基于深度學(xué)習(xí)算法和水藥效團(tuán)的seq2seq模型。
首先通過(guò)深度學(xué)習(xí)算法,設(shè)計(jì)出一系列滿足類藥五原則等諸多性質(zhì)的苗頭化合物,接著使用分子對(duì)接和藥效團(tuán)模型,評(píng)估這些苗頭化合物的有效性,其中的藥效團(tuán)模型用的是水藥效團(tuán)模型,這個(gè)模型通過(guò)水分子的分子動(dòng)力學(xué)模擬來(lái)構(gòu)建藥效團(tuán)特征,其中藥物靶點(diǎn)的水藥效團(tuán)特征如圖4所示。

圖4. 六種藥物靶點(diǎn)的水藥效團(tuán)特征,來(lái)源:參考文獻(xiàn)6
注:6 個(gè)藥物靶點(diǎn) (AR、GR、PR、PARP、AChE 和 PPAR ) 的水藥效團(tuán)特征。疏水特征,綠色球體;芳香特征,橙環(huán);氫鍵受體,紅色球體;氫鍵供體,藍(lán)色球體;特征容差,透明球體;排除體積的結(jié)合部位,藍(lán)色透明球體。
該方法可以自主產(chǎn)生一系列靶向有利的化合物,從大規(guī)模的化合物庫(kù)中篩選出匹配藥效團(tuán)特征的分子,以這些分子作為輸入,訓(xùn)練分子生成模型。最后用藥效團(tuán)篩選生成的分子,得到一批候選化合物。水藥效團(tuán)模型用于在大型化合物數(shù)據(jù)庫(kù)中,篩選對(duì)給定目標(biāo)有利的化合物,而seq2seq化合物生成器用于訓(xùn)練篩選出的化合物,并基于訓(xùn)練模型生成全新的化合物。
4解釋藥物中的構(gòu)效關(guān)系
DNN指的是由神經(jīng)網(wǎng)絡(luò)組成的、復(fù)雜的非線性統(tǒng)計(jì)模型[10],該網(wǎng)絡(luò)具有多個(gè)用于預(yù)測(cè)的隱含層(如圖5所示)。更快的計(jì)算機(jī)硬件、改進(jìn)的算法,以避免神經(jīng)網(wǎng)絡(luò)過(guò)度匹配,再加上許多計(jì)算機(jī)平臺(tái)上軟件解決方案的可用性以及改進(jìn)的訓(xùn)練算法,推動(dòng)了 DNN在人工智能領(lǐng)域的成功應(yīng)用,例如計(jì)算機(jī)視覺、語(yǔ)言處理以及藥物設(shè)計(jì)。
在計(jì)算機(jī)領(lǐng)域,基于深度神經(jīng)網(wǎng)絡(luò) (DNN) 的模型在預(yù)測(cè)新分子的活性和性質(zhì)方面很有前途。不幸的是,它們固有的黑箱特征阻礙了我們對(duì)分子結(jié)構(gòu)作用的理解。然而,這些信息對(duì)于研究構(gòu)效關(guān)系(SAR) 以指導(dǎo)進(jìn)一步優(yōu)化至關(guān)重要。
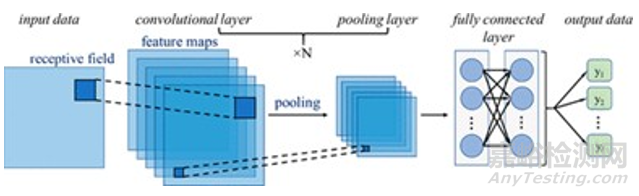
圖5. 卷積神經(jīng)網(wǎng)絡(luò) (CNN) 示意圖,來(lái)源:參考文獻(xiàn)10
為了解決這一解釋差異,“可解釋人工智能”(XAI) 方法最近變得流行起來(lái)。Tobias, Hans[7]等人將多種XAI方法應(yīng)用于SAR和X射線晶體結(jié)構(gòu)的先導(dǎo)優(yōu)化數(shù)據(jù)集的項(xiàng)目(如圖 6 所示),并進(jìn)行了比較,結(jié)果說(shuō)明將DNN模型與一些強(qiáng)大的解釋方法相結(jié)合,可以得到易于理解的、全面的解釋。

圖6. 解釋構(gòu)效關(guān)系的方法流程,來(lái)源:參考文獻(xiàn)7
這些解釋可以使用熱圖以一種全面的方式收集和呈現(xiàn),以清楚地突出分子的哪些部分被認(rèn)為對(duì)解釋親和力或任何其他目標(biāo)性質(zhì)有利或不利,這將激發(fā)計(jì)算化學(xué)家的創(chuàng)造力,以設(shè)計(jì)出具有前瞻性思維的藥物。
5未來(lái)改進(jìn)趨勢(shì)
方向合理的藥物設(shè)計(jì)方法正在不斷改進(jìn),這些方法正在接近更廣泛的藥物靶點(diǎn),未來(lái)還可以預(yù)計(jì)會(huì)有各種各樣的額外改進(jìn):
1. 改進(jìn)的計(jì)算機(jī)硬件允許使用更嚴(yán)格的方法應(yīng)用于大分子系統(tǒng),如果在未來(lái)看到量子力學(xué)對(duì)接研究的出現(xiàn)也并不令人驚訝,此外還會(huì)促進(jìn)混合方法的發(fā)展[2](遺傳神經(jīng)網(wǎng)絡(luò)、近鄰遺傳算法等)和藥物設(shè)計(jì)集成工具(設(shè)計(jì)與評(píng)估一體化)的開發(fā)。
2. 基于人工智能的蛋白質(zhì)靶點(diǎn)結(jié)構(gòu)、位點(diǎn)解析將會(huì)更加深入地滲透進(jìn)藥物設(shè)計(jì)中,即蛋白質(zhì)結(jié)構(gòu)建模的進(jìn)展,促進(jìn)了基于結(jié)構(gòu)的藥物設(shè)計(jì)在未結(jié)晶的藥物靶點(diǎn)上的廣泛使用,它們是化學(xué)設(shè)計(jì)的補(bǔ)充,并且在選擇用于風(fēng)險(xiǎn)評(píng)估的實(shí)驗(yàn)系統(tǒng)時(shí)很重要。例如:AO底物類藥物的設(shè)計(jì)[3](AO是醛氧化酶,結(jié)構(gòu)作用如圖7所示,包括催化氮雜雜環(huán)和醛的氧化、酰胺水解和多種還原),AO底物在上市藥物中很少見,許多候選藥物由于藥代動(dòng)力學(xué)活力差、種間差異和不良反應(yīng)而失敗。

圖7. 醛氧化酶的結(jié)構(gòu)及其結(jié)合位點(diǎn),來(lái)源:參考文獻(xiàn)3
由于大多數(shù)問(wèn)題源于復(fù)雜且知之甚少的AO生物學(xué)結(jié)構(gòu),因此有效的解決方案是停止或減少AO代謝:早期考慮AO介導(dǎo)的代謝(例如,在苗頭化合物和先導(dǎo)化合物優(yōu)化中),但不是主動(dòng)避免AO結(jié)構(gòu),其中許多是有價(jià)值的和常見的構(gòu)建塊(例如,各種氮雜雜環(huán)和酰胺),及時(shí)合理地應(yīng)用藥物設(shè)計(jì)來(lái)控制AO介導(dǎo)的代謝(例如,停止、減少或用于前藥設(shè)計(jì));
3. 與人工智能相結(jié)合的新實(shí)驗(yàn)方法將藥物設(shè)計(jì)引向新的方向,有許多類似的例子:如果沒(méi)有固相合成方法,組合化學(xué)[11](實(shí)例如圖 8 所示)和高通量篩選將不會(huì)像今天這樣有用;在催化劑設(shè)計(jì)等領(lǐng)域的改進(jìn),在催化劑設(shè)計(jì)等領(lǐng)域的改進(jìn),使我們可以快速訪問(wèn)更多的化學(xué)結(jié)構(gòu),使生物活性測(cè)定可以利用更廣泛的生物靶標(biāo)。

圖8. 組合化學(xué)實(shí)例,來(lái)源:參考文獻(xiàn)11
例如,結(jié)合人工智能和統(tǒng)計(jì)學(xué)方法對(duì)CADD的藥物評(píng)估算法本身進(jìn)行優(yōu)化[8],定量方法僅對(duì)結(jié)構(gòu)相似分子的抑制劑之間的相對(duì)結(jié)合親和力提供準(zhǔn)確預(yù)測(cè),而定性方法為結(jié)構(gòu)更多樣化的一組化合物的相對(duì)結(jié)合親和力提供定性趨勢(shì)。
理想情況下,結(jié)合這兩個(gè)特征將大大提高藥物評(píng)估算法在藥物設(shè)計(jì)中的效用(如圖9所示)。即結(jié)合人工智能和統(tǒng)計(jì)學(xué)方法計(jì)算出包含上述許多特性的回歸方程,然后將方程嵌入CADD的藥物評(píng)估算法,將極大有利于藥物設(shè)計(jì)。

圖9. 定性、定量方法對(duì)CADD的影響
結(jié)語(yǔ)
人工智能不會(huì)是藥物化學(xué)家的末日,但它會(huì)是不使用人工智能的藥物化學(xué)家的末日。藥物化學(xué)家自身設(shè)計(jì)的藥物的復(fù)雜度即將達(dá)到極限,漸凍癥、老年癡呆癥以及癌癥等復(fù)雜疾病始終無(wú)法被攻克就是最好的預(yù)兆,而將人工智能應(yīng)用于藥物設(shè)計(jì)將是解決這一問(wèn)題的鑰匙。并且醫(yī)藥領(lǐng)域的人工智能需要從計(jì)算化學(xué)的興衰中吸取教訓(xùn),避免從信任到不信任的災(zāi)難性衰退,最后才作為一個(gè)可靠而有用的工具從灰燼中重生的經(jīng)歷[9]。
總之,人工智能中的藥物設(shè)計(jì)是一個(gè)令人興奮且不斷發(fā)展的研究領(lǐng)域,它對(duì)生活質(zhì)量和健康的影響決定了該領(lǐng)域的活力。
參考資料:
[1] https://blog.benchsci.com/pharma-companies-using-artificial-intelligence-in-drug-discovery
[2] Reddy M R , Parrill A L . Overview of Rational Drug Design[M]. 1999.
[3] Manevski N , King L , Pitt W R , et al. Metabolism by Aldehyde Oxidase: Drug Design and Complementary Approaches to Challenges in Drug Discovery[J]. Journal of Medicinal Chemistry, 2019, 62(24).
[4] Krishnan S R , Bung N , Vangala S R , et al. De Novo Structure-Based Drug Design Using Deep Learning[J]. 2021.
[5] Alberga D , Gambacorta N , Trisciuzzi D , et al. De Novo Drug Design of Targeted Chemical Libraries Based on Artificial Intelligence and Pair-Based Multiobjective Optimization[J]. Journal of Chemical Information and Modeling, 2020.
[6] Kim M , Park K , Kim W , et al. Target-Specific Drug Design Method Combining Deep Learning and Water Pharmacophore[J]. Journal of Chemical Information and Modeling, 2021, 61(1):36-45.
[7] Harren, Tobias et al. “Interpretation of Structure-Activity Relationships in Real-World Drug Design Data Sets Using Explainable Artificial Intelligence.”Journal of chemical information and modeling vol. 62,3 (2022): 447-462.
[8] Miljkovi F , R Rodríguez-Pérez, Bajorath J . Impact of Artificial Intelligence on Compound Dis- covery, Design, and Synthesis[J]. 2021.
[9] Jordan A M . Artificial Intelligence in Drug Design—The Storm Before the Calm[J]. Acs Medicinal Chemistry Letters, 2018.
[10] Yang X , Wang Y , Byrne R , et al. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery[J]. Chemical Reviews, 2019, 119(18).
[11] Struble T J , Alvarez J C , Brown S P , et al. Current and Future Roles of Artificial Intelligence in Medicinal Chemistry Synthesis[J]. 2020.

來(lái)源:藥渡


