您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2021-08-19 12:35
• 藥物發現和研發是制藥企業和化學科學家的重要研究領域。然而,低效率和高成本給該領域帶來了障礙。此外,處理來自基因組學、蛋白質組學、微陣列和臨床試驗的大量復雜數據也存在挑戰。
人工智能和機器學習技術使制藥領域實現了現代化。機器學習和深度學習算法已被應用于多肽合成、虛擬篩選、毒性預測、藥物監測和釋放、藥效團建模、定量構效關系、藥物重定位、多藥理和生理活性等藥物發現過程。此外,新的數據挖掘和管理技術為最近開發的建模算法提供了支持。
今天為大家解讀的文獻是Molecular Diversity雜志于今年4月發表的《Artificial intelligence to deep learning: machine intelligence approach for drug discovery》[1],第一作者為印度DTU的Rohan Gupta教授。由于閱讀后對編者啟發很大,所以整理后分享出來,如有理解不當之處,懇請大家批評指正。

圖1 人工智能在藥物發現與研發中的應用
人工智能的演進:從機器學習到深度學習
人工智能(AI),也被稱為機器智能,指的是計算機系統從輸入或過去的數據中學習的能力,術語“人工智能”通常用于機器在學習和解決問題過程中模仿與人腦相關的認知行為。2015年9月,谷歌顯示AI已成為搜索量最大的詞匯。
一些人將機器學習(ML)描述為“AI的應用”,而另一些人則將其描述為“AI的子集”。ML將數據與樸素貝葉斯、決策樹(DT)、隱馬爾可夫模型(HMM)等算法一起輸入機器,使其在不顯式編程的情況下進行學習。大約在20世紀中旬,Igor Aizenberg和他的同事們在談論人工神經網絡(ANN)時,首次提出了“深度學習(DL)”這一術語。
根據《人工智能:現代方法》中的討論,人工智能有七種分類(圖2),分別是推理和問題解決、知識表示、規劃和社會智能、感知、機器學習、機器人:運動和操縱,以及自然語言處理。機器學習進一步分為三個重要子集:監督學習、無監督學習和深度學習;而自然語言處理被分為五個主要子集,包括分類、機器翻譯、問答、文本生成和內容提取。可以說,DL是ML的子集,ML是AI的子集,進化的順序是AI>ML>DL。

圖2 人工智能的分類
藥物發現的革命性過程:大數據和人工智能的作用
大數據可以定義為過于龐大和錯綜復雜的數據集,無法使用傳統的數據分析軟件、工具和技術進行分析。大數據的三個主要特征是體積、速度和多樣性,其中體積代表產生的大量數據,速度代表這些數據被再現的速率,多樣性代表數據集中存在的異質性。隨著微陣列、RNA-seq和高通量測序(HTS)技術的出現,每天都會產生過多的生物醫學數據,當代藥物發現也因此進入了大數據時代。
在藥物發現中,第一步也是最重要的一步是確定與疾病病理生理學有關的適當靶點(如基因、蛋白質),然后找到可以干擾這些靶點的藥物或類藥物分子。如今我們可以搜索一系列生物醫學數據庫來實現,如NCBI GEO、癌癥基因組圖譜(TCGA)和Arrayexpress等等。有時甚至出版的文獻也可以用于識別靶點,如PubMed是各種已出版生物醫學文獻的數據庫,對其進行數據挖掘可以幫助識別不同疾病的靶點。此外,人工智能的發展使得大數據分析變得容易得多,因為現在有無數的ML技術可用,這些技術可以幫助提取這些大型生物醫學數據集中存在的有用特征、模式和結構。如Han等人[2]利用大數據和人工智能在2019年開發了DriverML,這是一個基于ML監督學習的工具,可以指出與癌癥相關的驅動基因。
在確定和驗證了合適的靶點之后,下一步是尋找合適的藥物或類藥物分子,這些分子可以與靶點相互作用并引起所需的反應。在大數據時代,我們可以支配海量的大型化學數據庫,這些數據庫可以幫助我們找到針對特定靶點的完美藥物。比如PubChem是一個免費的化學數據庫,其中包含各種化學結構的數據,包括它們的生物、物理、化學和毒性特性;ChEMBL包含許多具有類似藥物特性的生物活性化合物的數據,還包含有關這些化合物的吸收、分布、新陳代謝和排泄(ADME)、毒性特性,甚至它們的靶相互作用的信息;其他的化學數據庫還包括DrugBank、LINCS L1000和PDB等。

圖3 大數據在藥物設計和發現中的應用
人工智能和傳統化學的結合:促進藥物發現
隨著技術的進步和高性能計算機的發展,在計算機輔助藥物設計(CADD)中補充了從ML到DL的一系列人工智能算法。在過去的二十年里,發展了許多用于計算藥物發現、定量結構活性關系(QSAR)和自由能最小化技術的工具。例如,使用機器智能方法(如DT、隨機森林(RF)方法、CNN、SVM、LSTM網絡和梯度增強機)區分復合細胞活性。類似地,使用QSAR方法預測PAMPA有效滲透率時,結果表明相比于偏最小二乘(PLS)方案,使用分層支持向量機(HSVR)方案開發的基于ML的模型在訓練集、測試集和統計分析方面執行得更好。另外,對于新化合物的合成,化學科學家常常借助已發表的文獻,而隨著涉及AI和ML的自動藥物發現方法的進步,區分現有藥物和新的化學結構變得相對簡單。
傳統的面向化學的藥物發現與人工智能藥物設計相結合,提供了一個很好的研究平臺。此外,世界各地的系統生物學和化學科學家與計算科學家合作,開發現代ML算法和原理,可以促進藥物的發現和開發。
人工智能在藥物發現與藥物研發中的應用
在藥物發現和開發過程中,最艱巨和令人沮喪的一步是尋找存在于浩瀚化學空間中合適的、具有生物活性的藥物分子;而最令人氣憤的,是十分之九的藥物分子通常不能通過第二階段臨床試驗和其他監管批準。上述事件可以通過實施基于人工智能的工具和技術來解決。人工智能可以參與藥物開發過程的每一個階段:
5.1 一次和二次藥物篩選
在藥物發現中,先導化合物的篩選是至關重要的,人工智能在識別新的和潛在的先導化合物方面發揮著巨大的作用。在化學空間中有大約1.06億個化學結構,他們來自不同的研究,如基因組研究、臨床和臨床前研究、體內分析和微陣列分析。利用機器學習模型,如強化模型、Logistic模型、回歸模型和生成模型,根據活性位點、結構和靶結合能力可以篩選出這些化學結構。

圖4 人工智能在一次和二次藥物篩選中的應用
5.2 肽合成與小分子設計
多肽是一種由大約2-50個氨基酸組成的生物活性小鏈,由于它們具有跨越細胞屏障的能力并可以到達所需的靶點,因此越來越多地被用于治療。近年來,研究人員利用人工智能的優勢發現了新肽。例如,Yan[3]等人在2020開年發了基于DL的短抗菌肽(AMPs)鑒定平臺Deep-AmPEP30。AmPEP30是一種CNN驅動的工具,可以根據DNA序列數據預測短AMP。使用該平臺,研究人員從一種存在于胃腸道的真菌病原體——光滑梭菌的基因組序列中鑒定出新的AMPs。
小分子是分子量非常低的分子,就像肽一樣,利用人工智能也可以用來探索小分子的治療作用。例如,Zhavoronkov等人[4]設計了一種基于生成性強化學習的小分子從頭設計工具GENTRL,并利用它發現了一種新的酶抑制劑,DDR1激酶。
5.3 藥物劑量和給藥效果的識別
給病人任何不適當劑量的藥物都可能導致不良和致命的副作用,多年來,確定能夠以最小毒副作用達到預期效果的藥物的最佳劑量一直是一個挑戰。隨著人工智能的出現,許多研究人員正在借助ML和DL算法來確定合適的藥物劑量。
例如,Shen等人[5]開發了一個基于人工智能的平臺,稱為AI-PRS,用于確定通過抗逆轉錄病毒療法治療艾滋病毒的最佳劑量和藥物組合。AI-PRS是一種神經網絡驅動的方法,它通過拋物線響應曲線(PRS)將藥物組合和劑量與療效聯系起來。在他們的研究中,10名HIV患者聯合使用替諾福韋、法韋倫和拉米夫定,AI-PRS分析表明替諾福韋的劑量可以減少起始劑量的33%,而不會導致病毒復發。
5.4 生物活性物質預測與藥物釋放監測
最近已經開發了多種在線工具來分析藥物釋放,以及選定的生物活性化合物作為載體的可行性。最常用的是基于化學特征的藥效團評價。為了研究基于配體的化學性質,已經使用CATALYST程序建立了各種成功的實驗。此外,利用人工智能研究人員可以確定用于與疾病相關的特定靶點的生物活性化合物。例如,Wu等人利用集成DL和RF的方法設計了WDL-RF用于測定靶向配體的G蛋白偶聯受體(GPCRs)的生物活性。
5.5 蛋白質折疊和蛋白質相互作用的預測
分析蛋白質-蛋白質相互作用(PPI)對于藥物開發和發現至關重要。比如使用貝葉斯網絡(BN)預測PPI,其本質是利用基因共表達、基因本體(GO)和其他生物過程相似性,集成數據集產生精確的PPI網絡。已有研究小組使用BN結合酵母菌的數據集研究出一種新的層次模型PCA集成極限學習機(PCA-EELM),該工具可以僅使用蛋白質序列信息來預測蛋白質-蛋白質相互作用,提供準確且快速的輸出[6]。
5.6 基于結構和基于配體的虛擬篩選
在藥物設計和藥物發現中,虛擬篩選(VS)是CADD的重要方法之一,是從化合物庫中篩選出有前景的治療化合物的有效方法。作為高通量篩選的重要工具,它也帶來了成本高、準確率低的問題。要將ML用于VS,應該有一個由已知的活性和非活性化合物組成的過濾訓練集。這些訓練數據用于使用監督學習技術訓練模型。然后對訓練的模型進行驗證,如果它足夠精確,則將該模型用于新的數據集,以針對目標篩選具有所需活性的化合物。ML能夠加快VS的速度,使其更完善,甚至可以減少VS中的誤報。
一般來說VS分兩種,基于結構的VS(SBVS)和基于配體的VS(LBVS)(圖5)。其中,分子對接是SBVS中應用的主要原則,已經開發了幾種基于AI和ML的評分算法,如NNScore、CScore、SVR-SCORE和ID-SCORE;也有算法被開發用于SBVS中的分子動態模擬分析以及預測SBVS中蛋白質-配體的親和力,如RFS、支持向量機、CNNs和淺層神經網絡。類似的, LBVS也開發了不同的算法和工具,例如SwissSimilarity、METADOCK、HybridSim-VS、PKRank、BRUSELAS和AutoDock Bias等等。

圖5 基于配體的(A)和基于結構的(B)虛擬篩選
5.7 QSAR建模與藥物再利用
在藥物設計和開發中,研究化學結構和理化性質與生物活性之間的關系是至關重要的。定量構效關系(QSAR)建模是一種計算方法,通過它可以在化學結構和生物活性之間建立定量的數學模型。傳統QSAR模型大致分為兩類,回歸模型(如高斯過程GPs)和分類模型。目前已經開發了多種基于網絡的工具和算法,如Vega平臺、QSAR-Co、FL-QSAR、Transformer-CNN和Chemception等,為QSAR建模提供了一條新的途徑。
在藥物設計和發現中,藥物重新定位是指對已經針對一種疾病情況開發的藥物進行調查,并針對其他疾病情況進行重新定位。近年來,基于人工智能的工具和算法的出現為該領域研究提供了平臺,如DrugNet、DRIMC、DPDR-CPI、PHARMGKB和DRRS等。特別是最近,新冠肺炎成為一種全球性的流行病,世界各地的研究人員開始尋找有前途的治療劑。在這方面,Hooshmand等人基于神經網絡進行藥物重新定位,確定了16種潛在的抗HCoV可再利用藥物,并為新冠肺炎確定了12個有前景的藥物靶點[7]。
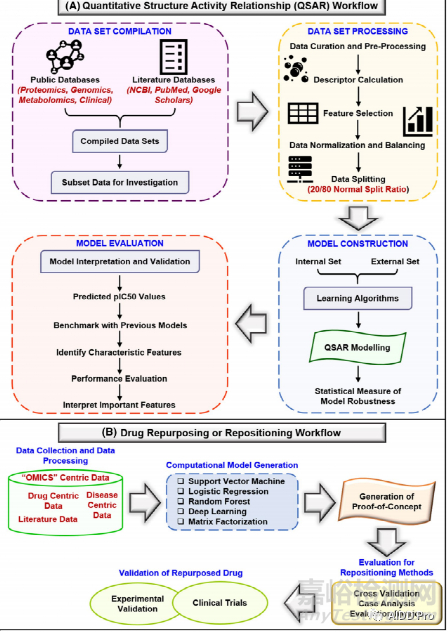
圖6 QSAR建模(A)與藥物再利用(B)
5.8 理化性質和生物活性的預測
眾所周知,每一種化合物都與溶解度、分配系數、電離度、滲透系數等物理化學性質有關,這可能會阻礙化合物的藥代動力學特性和藥物靶向結合效率。因此,在設計新的藥物分子時,必須考慮化合物的物理化學性質。為此,已經開發了不同的基于人工智能的工具來預測這些性質,包括分子指紋、SMILES格式、庫侖矩陣(Coulomb matrices)和勢能測量,這些都用于DNN訓練階段。
此外,藥物分子的治療活性取決于其與受體或靶點的結合效率,因此,預測化學分子與治療靶點的結合親和力對于藥物的發現和開發至關重要。人工智能算法的最新進展增強了該過程,使用相似性特征已經開發了幾個基于網絡的工具,如ChemMapper和相似集合方法(SEA)。此外,還構建了基于ML和DL的藥物靶標親和力識別模型,如KronRLS、SimBoost、DeepDTA和Padme等。
5.9 化合物的作用方式和毒性預測
藥物毒性是指化學分子由于化合物的作用方式或新陳代謝方式而對生物體產生的不利影響。人工智能可以預測藥物分子與靶點結合和未結合時的效應,以及體內安全性分析。已經開發了不同的基于Web的工具,如LimTox、pkCSM、admetSAR和Toxtree。
5.10 分子通路的鑒定與多重藥理學
人工智能和最大似然算法在藥物發現和開發中的重要成果之一是預測和估計疾病網絡、藥物-藥物相互作用和藥物-靶點關系的總體拓撲和動力學。數據庫如DisGeNET、STRTCH、STRING分別被用于確定基因-疾病關聯、藥物-靶標關聯和分子途徑。例如,Gu等人在2020年使用相似性集成方法確定了197種最常用中草藥的靶點,然后使用DisGeNET數據庫將這些靶標與不同的疾病聯系起來,從而將草藥與可用于治療的疾病聯系起來[8]。
在藥物化學中,多重藥理學是指在與疾病相關的藥物靶標生物網絡中設計能夠與多個靶點相互作用的單一藥物分子。它適合于為復雜疾病,如癌癥、神經退行性疾病(NDDS)、糖尿病和心力衰竭等設計治療劑。由于強大的挖掘能力和數據分析能力,基于ML的方法具有分析牽連分子網絡的潛力,大大增加發現多靶配體的概率。此外,ML模型有助于識別具有不同結合口袋的多靶配體。
5.11 臨床試驗的設計
在引入人工智能技術后,臨床試驗的成功率大幅提高。IBM Watson開發了一個臨床試驗配對系統[9],該系統使用患者的醫療記錄和大量過去的臨床試驗數據來創建詳細的檔案。人工智能模型還可以通過分析毒性、副作用和其他相關參數來提高成功率,從而降低臨床試驗的成本。
未來的挑戰和可能的解決方案
目前,制藥行業在開發新藥時面臨的主要挑戰是成本高和效率低。ML方法和DL的最新發展帶來了“降本增效”的巨大機會,這也引起了各地研究人員的極大興趣,以至于許多制藥公司都與人工智能公司合作。此外,該領域的初創公司數量也在不斷攀升,到2020年6月達到了230家。
如今,人工智能在藥物發現領域被用于靶標識別、先導優化、ADME-T預測和構建臨床試驗等各個步驟。盡管取得了巨大的成功,但仍然存在許多挑戰,其中有兩個最重要的問題:首先,標記不能是二元的,因為藥物在生物系統中的作用是復雜的;其次,雖然數據庫擁有海量信息,但藥物發現中可用的高質量數據并不多。因此,需要一個不僅能提供數據數量而且能提供質量的平臺。在制藥行業,開放數據共享并不常見,皮斯托亞聯盟(Pistoia alliance)主動發起了一場運動,鼓勵許多公司與他人共享數據,在未來他們還打算建立統一的數據格式。
2020年12月,In silico medicine公司為他們的小分子抑制劑向FDA提出了IND申請,他們的目標是在2022年初進行臨床試驗。如果試驗成功,那么這將是有史以來第一次通過基于人工智能的工具提出并批準一種新的靶點及其抑制劑。盡管將人工智能工具融入藥物發現過程中存在一些不可避免的障礙,還有大量的工作要做,但毫無疑問,在不久的將來,人工智能將給藥物發現和開發過程帶來革命性的變化。
參考文獻
[1] Gupta, R., Srivastava, D., Sahu, M. et al. Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Mol Divers (2021)
[2] Han Y, Yang J, Qian X et al (2019) DriverML: a machine learning algorithm for identifying driver genes in cancer sequencing studies. Nucleic Acids Res.
[3] Yan J, Bhadra P, Li A et al (2020) Deep-AmPEP30: improve short antimicrobial peptides prediction with deep learning. Mol Ther-Nucleic Acids 20:882–894.
[4] Zhavoronkov A, Ivanenkov YA, Aliper A et al (2019) Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 37:1038–1040.
[5] Shen Y, Liu T, Chen J et al (2020) Harnessing artificial intelligence to optimize long-term maintenance dosing for antiretroviral-naive adults with HIV-1 Infection. Adv Ther 3:1900114.
[6] You ZH, Lei YK, Zhu L et al (2013) Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinformatics 14:1–11.
[7] Hooshmand SA, Zarei Ghobadi M, Hooshmand SE et al (2020) A multimodal deep learning-based drug repurposing approach for treatment of COVID-19. Mol Divers.
[8] Gu S, Lai L, hua, (2020) Associating 197 Chinese herbal medicine with drug targets and diseases using the similarity ensemble approach. Acta Pharmacol Sin 41:432–438.
[9] Fogel DB (2018) Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: a review. Contemp Clin Trials Commun 11:156–164.

來源:AIDD Pro


