您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2020-09-16 18:05
關于專屬性,中國藥典給出的定義是:在其他成分存在的條件下,采用的分析方法能夠正確測出被測物的能力。鑒別反應、雜質檢查和含量測定方法均應考察專屬性。藥典中就鑒別反應、雜質檢查和含量測定的專屬性均做出了詳細說明。在實際驗證過程中,大多數情況下我們會按部就班去做。但有一些方法,其屬于非專屬性方法,如滴定法,其專屬性只可以通過其他方法去佐證。本文就專屬性的實際操作及相關問題進行簡單論述,不足之處還請指正。
一、專屬性的具體做法討論
本文主要就雜質檢查和含量測定方法的專屬性做法進行論述。
1、含量測定
藥典描述:對于含量測定,在雜質對照品可獲得的情況下,可在試樣中加入雜質或輔料,考察測定結果是否受干擾,并與未加入雜質或輔料的試樣比較測定結果。在雜質或降解產物不能獲得的情況下可用另一個經驗證了的方法或藥典方法比較結果,也可用強光照射、高溫、高濕、酸(堿)水解或氧化等方法進行加速破壞,以研究可能存在的降解產物對含量測定的影響。含量測定應對比兩種方法的結果。
在藥典和ICH中,含量測定與雜質檢查的專屬性沒有明確的分開描述,所以實際中大部分情況下含量測定與雜質檢查的專屬性做法基本保持一致。這里有以下幾點需要考慮:
(1)含量測定方法與雜質檢查方法完全相同。
在此情況下,既需要考察雜質能夠準確測定,又需要考察含量測定的準確性。由于供試品濃度完全一致,在雜質和輔料存在的情況下,需要著重考察雜質和輔料與主成分的分離情況以及雜質與輔料、雜質與雜質間的分離情況,對于含量測定,一般要求雜質和輔料均能與主成分有效分離。其次是進行破壞試驗,一般要求有一種或幾種破壞能夠得到相應的降解產物,觀察降解產物與主成分峰的分離度,并進行峰純度檢查。在含量測定方法與雜質檢查方法完全相同的情況下,以上做法是最常見的,但這種做法忽略了考察含量測定本身的準確性,僅考察了主成分不受干擾的情況,忽略了測定的重點的含量。
(2)含量測定與雜質檢查方法色譜條件相同,但供試品濃度不同。
在此種條件下,往往是含量測定供試品濃度遠低于雜質檢查的供試品濃度。首要考慮的是,當供試品濃度較低時,雜質和輔料在該色譜條件下是否會出峰。具體的做法:使用與主成分相鄰雜質對照品按照限度的120%或150%配制相應濃度的溶液,在該色譜條件下進樣,觀察出峰情況。按照處方量配制相應的輔料溶液,在該色譜條件下進樣,觀察出峰情況。這里涉及到含量測定供試品濃度選擇的問題,一般會選擇限度的120%或150%的雜質不出峰對應的濃度作為含量測定供試品濃度。在此種情況下,考察主成分與雜質或輔料的分離情況意義不大,降解試驗也不適用。
(3)含量測定與雜質檢查方法完全不同。此種條件下具體實施方法需要根據供試品濃度的選擇情況而定。
如以上所述,三種情況下均需要考慮含量測定的準確性。就外標法測定含量來講,需要考察在雜質和輔料存在的情況下含量測定的結果不受影響。具體的操作如下:
按照含量測定供試品濃度,使用原料與限度濃度的相鄰雜質配制成溶液1;
按照含量測定供試品濃度,使用原料與處方量輔料配制成溶液2;
向供試品中加入限度濃度的雜質,配制成溶液3;
供試品溶液作為對照溶液。
分別取溶液1、2、3和對照溶液測定含量,對比含量測定結果,要求結果應無明顯差異。取供試品進行適量破壞試驗,配制成相應的供試品溶液,進行含量測定,測定結果與未破壞含量結果應無明顯差異。這里所說的適量破壞試驗,降解產生1%~2%的雜質即可,破壞程度較強,此種對比就會失去原本的意義。
2、雜質檢查
藥典描述:在雜質對照品可獲得的情況下,對于雜質檢查,可向試樣中加入一定量的雜質,考察各成分包括雜質之間能否得到分離。在雜質或降解產物無法獲得的情況下,可用另一個經驗證了的方法或藥典方法比較結果,也可用強光照射、高溫、高濕、酸(堿)水解或氧化等方法進行加速破壞,雜質檢查應對比檢出的雜質個數,必要時可采用光二極管列陣檢測器和質譜檢測進行峰純度檢查。
對于雜質檢查的專屬性,一般做法:按照雜質檢查供試品濃度,使用原料和限度濃度的雜質配制成混合溶液;取各雜質配制成各單個雜質溶液;按照處方量取輔料混合制成輔料溶液;分別取空白溶劑、混合溶液、各單個雜質溶液和輔料溶液進樣,進行相應峰的定位,以說明各雜質峰、輔料峰、空白溶劑峰和主成分峰的分離情況。取供試品進行破壞試驗,觀察降解產物與主成分峰的分離情況,使用光二極管列陣檢測器進行峰純度檢查。個人認為在應當使用光二極管列陣檢測器和質譜檢測兩種方法同時進行峰純度檢查。
二、專屬性相關的一些問題
1、雜質檢查專屬性中破壞試驗的破壞程度
關于破壞試驗的破壞程度,目前較為廣泛認可的是5%~10%。個人認為尚不能如此定論,具體從以下幾點進行論述:
(1)需要考慮方法建立之初雜質能夠分離的承載量,即雜質與雜質之間、主峰與相鄰雜質之間能夠有效分離時的最大濃度。
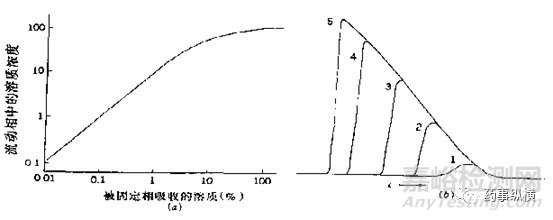
圖1 樣品量對峰寬與峰形的影響
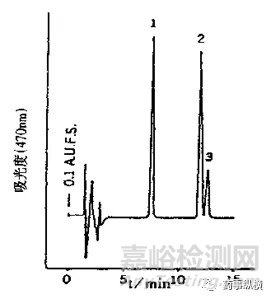
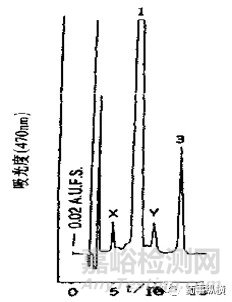
圖2(上)、3(下)雜質與雜質之間、主峰與雜質之間的分離情況
如圖1,列出了樣品量對峰寬和峰形影響的一種情況。當溶液中樣品量增加時,峰寬會相應變寬。如圖2(上)所示,雜質2與雜質3在相應限度的情況下可以達到基本分離,但若破壞試驗時雜質2與雜質3的量超過限度,則會因峰寬變寬影響二者之間的分離度。如圖3(下)所示,雜質Y與主峰1之間完全分離,但若破壞試驗時雜質Y的量超過限度很多(如雜質Y達到0.5%),則會由于其峰寬變寬影響與主峰的分離度,導致主峰峰純度達不到要求。
(2)藥物穩定性本身的考慮。
對藥物進行加速破壞試驗,一是為了了解藥物的降解途徑及相應的降解產物,二是為了考察降解產物在該分析方法下能不能被準確檢出。一般情況下,光照破壞采用可見光及近紫外條件下室溫放置1~3天,酸堿破壞最高使用1mol/L強酸強堿室溫條件放置1天,氧化破壞最高使用30%過氧化氫溶液室溫放置1 天,高溫可采用60~80℃最長放置5天,便可知曉藥物在相應條件下的穩定性;選用更加苛刻的條件進行破壞并沒有多大意義,需要結合實際中遇到的情況。
(3)、與物料平衡之間的相互關系。
計算物料平衡,是為了判斷使用該檢測方法能否將產生的降解雜質全部準確檢出,破壞程度較強會直接影響物料平衡。
2、物料平衡的可接受范圍
目前所接觸到的一些資料中,物料平衡的可接受范圍基本定為90%~110%,但這在藥典中并沒有明確的規定。個人認為需要就實際情況而論。影響物料平衡的因素有以下幾個:
(1)雜質的響應因子
在進行分析方法驗證時,我們需要得到雜質的相對校正因子,一般會存在有雜質相對校正因子為0.9以下或1.1以上的,表明雜質在方法中的響應差異較大,同樣也會存在未知雜質響應差異較大的情況。當進行加速破壞試驗后,由于響應差異較大的原因,雜質的峰面積不能與主峰失去的峰面積劃等號,因此物料平衡算出來就不是100%。
(2)有未檢出的雜質
當破壞程度較為劇烈時,有可能會產生方法檢不出的雜質,其可能為生成的氣體、無機鹽、在該條件下沒有響應的物質、由于檢測時間較短在規定時間內無法出峰的物質等。由于破壞后失去了該部分物質,導致物料平衡算出來不是100%。
(3)雜質的范圍
在相應的濃度范圍內,濃度與響應值呈線性關系(或其他非線性模型),當超出該范圍,響應值會明顯不符合該種關系。若破壞程度較為劇烈,有雜質的響應值超出了其相應范圍內的某種特定關系,也會導致物料平衡算出來不是100%。
以上是列出的其中三種可能存在的因素,也可能存在其他因素。
驗證中的項目相互之間均有聯系,如在歸納物料平衡計算時結果不是100%的原因時,雜質的校正因子和線性范圍便可以進行相應的解釋。

來源:藥事縱橫


