您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2019-09-10 10:52
摘要:本文在梳理國際人用藥品注冊技術協調會(ICH)發布的《化學藥品的研發與生產》(Q11)及其問答文件(階段4)相關要求的基礎上,結合日常藥學審評工作以及具體案例分析,就化學合成原料藥中起始原料的選擇問題及質控要求展開初步探討。
關鍵詞:化學合成原料藥 起始原料 ICH Q11 案例分析
起始原料(Starting Material)既是原料藥注冊申請通用技術文件(CTD格式)中工藝描述的出發點,也是藥品生產質量管理規范(Good Manufacturing Practice,GMP)第一次實施的引入點,還是今后藥品生命周期管理(Lifecycle Management,ICH Q12)的起始點。對于化學合成原料藥而言,起始原料的選擇是一個首要的問題,也是原料藥藥學審評的重點及難點。
丁恩峰等總結了2006-2011年歐洲藥品質量理事會(EDQM)公布的化學實體原料藥首次申請歐洲藥典適應性(CEP)證書的十大缺陷,結果起始原料及其相關問題在所有缺陷問題中所占比例約為三分之一[1]。EDQM還曾于2016年12月公布了2015年下半年至2016年上半年20份首次申請化學實體原料藥CEP證書的十大缺陷[2],其中,起始原料不被監管機構所接受位列缺陷第四條,而缺陷第二條和缺陷第三條也和起始原料有關(雜質譜、制備工藝等)。由此可見,起始原料的選擇需引起申請人足夠的重視。
隨著國際人用藥品注冊技術要求協調會(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)發布的《原料藥開發與制造》(Q11)的正式實施,ICH國家/地區對于起始原料的選擇原則基本達成了共識。考慮到我國已正式加入ICH,藥品監管理念和技術要求也有待和國際現行通用技術要求接軌。筆者嘗試在進一步梳理ICH Q11問答文件正式稿(Questions and Answers, Step 4,2017-08- 23)[3]和原國家食品藥品監督管理總局(以下簡稱總局)“關于發布化學藥品新注冊分類申報資料要求(試行)的通告”(2016年第80號)[4]的相關要求的基礎上,結合日常藥學審評工作,就化學合成原料藥中起始原料的選擇問題及質控要求展開初步探討,以期為國內原料藥注冊申報提供一定的借鑒和幫助。
1 國內外相關要求
鑒于起始原料選擇的重要性和復雜性,各國及ICH均出臺了一系列的指導原則,國內已有文獻進行了相應的歸納與總結[5-8]。同時,總局發布的“關于發布化學藥品新注冊分類申報資料要求(試行)的通告”(2016年第80號)及其解讀[9],也進一步明確了我國化學仿制藥申報資料中對于起始原料的相關要求。其中以4、5.2類申報資料為例,要求提供起始原料的選擇依據,明確對終產品質量有明顯影響的關鍵步驟均應納入本品的生產工藝中。應根據從源頭開始全程控制藥品質量的要求,選擇合適的起始原料,起始原料的選擇應符合ICH Q11及歐盟的相關技術要求。對于外購的起始原料,需要提供起始原料生產商出具的制備工藝,并根據相關技術指導原則、技術要求對雜質進行全面的分析和控制,制訂合理的內控標準,說明內控標準(尤其是雜質限度與含量)的制定依據,并提供數批外購起始原料的質檢報告與相關圖譜等。對外購的關鍵起始原料應制定供應商審計計劃,并提供審計報告。
根據ICH Q7[10],起始原料是用于合成原料藥的原材料、中間體或本身即是原料藥。起始原料應是原料藥的重要結構片段,其可以是商品,外包定制或是自行合成,通常應有明確的化學性質和結構。需要指出的是ICH Q7強調了起始原料的監管地位,明確了起始原料是GMP開始正式實施的引入點,但并沒有明確起始原料的選擇依據。ICH Q11[11]進一步明確了起始原料選擇的6條原則,體現了基于科學和風險的監管思考,ICH Q11同時強調應綜合考慮所有的原則,還要求進一步論述起始原料選擇的合理性。ICH Q11雖沒有明確擬定工藝應包含幾步化學合成步驟,但提出通常應包含對原料藥雜質譜產生影響的步驟。同時,ICH Q11還明確若采用市售化學品作為起始原料,則可以不必額外論述選擇依據,但并未進一步明確市售化學品和定制化學品的區別。
考慮到ICH Q11指導原則的概況性以及實際情況的復雜性,ICH于2017年8月發布了ICH Q11問答文件正式稿[3],就起始原料的選擇問題及質控要求細化為16個系列問答,進一步闡述了相關原則。筆者現節選部分重要內容概況如下。
重要的結構片斷僅用于區分起始原料和一般試劑、溶劑、催化劑或其他原材料,僅單獨將重要結構片斷作為選擇起始原料的依據不被官方接受。
明確了雜質含量水平對原料藥雜質譜有影響的定義,以及ICH Q11例4中“持續存在”雜質的定義。說明對于工藝上游中早期形成的,經多步步驟后仍“持續存在”的雜質,可以基于一定的科學認知和風險評估,制訂相應的控制策略,并非必需要將此雜質的形成步驟納入生產工藝中。
明確了如何確定哪些工藝步驟會影響原料藥的致突變性雜質譜。同時,并不建議將涉及致突變性雜質的所有步驟、或涉及區域選擇性/立體選擇性的所有步驟均包括在工藝描述中。
明確了評估是否包括足夠工藝步驟所應考慮的事項,同時建議應保證在GMP條件下能有足夠的工藝步驟,以降低潛在交叉污染或是今后起始原料相應變更(如,合成路線、供應商)所帶來的風險。
明確了市售化學品(Commercially Available Chemical)和定制化學品(Custom Synthesized Chemical)的區別,已有多個供應商不能作為判斷市售化學品的唯一依據,并進一步就市售化學品的生產規模和結構特點進行了相關闡述。若起始原料不是市售化學品,則應提供其制備路線,以及起始原料中實際/潛在存在的雜質信息。明確應提供起始原料的質量標準,并論述擬定起始原料內控標準在原料藥整體控制策略中所起的作用。同時強調,申請人應對所有的起始原料進行適當的質量控制。
ICH Q11問答文件還闡述了關于起始原料的生命周期管理,以及起始原料的批準后變更管理,并提供了起始原料選擇的決策樹。
2 案例分析
筆者嘗試結合具體案例分析,進一步就ICH Q11的相關原則進行闡述,但需要指出的是,本研究觀點僅代表作者個人的現有認知,不代表任何監管機構的法規要求,同時也歡迎業界同仁的批評指正。另外,同ICH Q11所聲明的事項一致,本研究所列舉案例并不暗示其中涉及的起始原料或反應步驟(數目)可被接受。
2.1 關鍵結構單元
ICH Q11明確了起始原料應包括原料藥重要的結構片段。但是,重要的結構片斷僅用于區分起始原料和一般試劑、溶劑和催化劑,僅依據重要的結構片斷作為起始原料的選擇依據不被接受。
如圖 1所示,已有文獻公開了塞來昔布(Ⅴ,Celecoxib)的一種合成方法[12-14]。資料顯示,對甲基苯乙酮(Ⅰ)和三氟乙酸乙酯(Ⅳ)在堿性條件下經克萊森縮合(Claisen Condensation)得1,3-二酮(Ⅱ)中間體,后與對磺酰胺基苯肼(Ⅲ)經環合反應而得塞來昔布。
圖 1 塞來昔布案例分析-關鍵結構單元

部分申請人僅依據原料藥重要的結構片段作為選擇依據,擬以1, 3-二酮(Ⅱ)為起始原料,并僅在成品中對雜質A進行控制。考慮到1, 3-二酮僅經一步化學合成即得塞來昔布,工藝步驟過短,則對于潛在雜質的清除能力可能有限,同時僅通過成品放行檢驗不利于藥品整體質量控制,故建議基于生產工藝的理解和風險評估的結果,綜合考慮ICH Q11相關原則,合理選擇起始原料。同時,對甲基苯乙酮中還可能潛在殘留鄰甲基苯乙酮(Ⅶ),也應關注其相應的衍化及去除。需要指出的是,塞來昔布EP標準中并未收載鄰甲基苯乙酮的相應衍生物。若申請人參照歐洲藥典標準擬定成品有關物質檢查法,未考察鄰甲基苯乙酮及其衍生物的殘留情況,考慮到尚不明確歐洲藥典方法對于該雜質的檢出能力,故認為申報資料的雜質譜分析尚不全面。
2.2 工藝步驟涉及致突變性或異構體雜質
ICH Q11在申報資料3.2.S.2.2部分的工藝描述中,通常應該包括對原料藥的雜質譜產生影響的工藝步驟,但是,并不建議將涉及致突變性雜質的所有步驟、或涉及區域選擇性/立體選擇性的所有步驟均包括在生產工藝中。
如圖 2所示,結合文獻報道[15-16],塞來昔布合成工藝中涉及潛在致突變性雜質的步驟包括對磺酰胺基苯肼(Ⅲ)的合成步驟(對磺酰胺基苯肼本身即為潛在致突變性雜質)和三氟乙酸乙酯(Ⅳ)的合成步驟。其中,對磺酰胺基苯肼(Ⅲ)一般是由對氨基苯磺酰胺經亞硝酸鈉氧化后,酸性還原而得。經進一步檢索(如Scifinder),對氨基苯磺酰胺主要有兩條制備路線:硝基苯途徑和氯苯途徑。同時,三氟乙酸乙酯一般由乙醇和三氟乙酸/三氟乙酸酐經酯化反應而得,反應的催化劑包括濃硫酸或對甲苯磺酸等,可能潛在生成硫酸二烷基酯或對甲苯磺酸二烷基酯。一般認為,硝基苯、氯苯、硫酸二烷基酯和對甲苯磺酸二烷基酯均為含有警示結構的潛在致突變性雜質。
圖 2 塞來昔布案例分析-潛在致突變性雜質
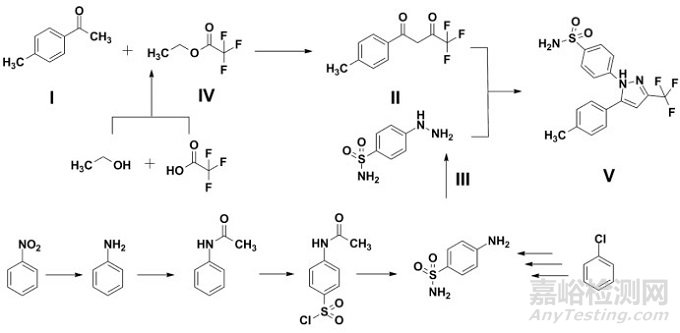
ICH Q11雖未強制要求將所有涉及產生致突變性雜質的步驟均包括在擬定工藝中,但是,建議結合ICH Q11和ICH M7[17](Genotoxic Impurities)指導原則的相關要求,全面評估原料藥的合成過程中所用的試劑、溶劑、中間體(包括合成原材料)以及潛在衍生物等的致突變性,基于對合成工藝的研究(包括已有文獻報道、研發數據積累等),明確工藝中實際/潛在存在的致突變性雜質,評估其形成步驟對于原料藥致突變性雜質譜是否存在影響,對于各潛在致突變性雜質建立相應的控制策略,同時保證在GMP條件下應有足夠的反應步驟,在此基礎上,合理選擇起始原料,以保證患者用藥安全。另需要指出的是,圖 2中塞來昔布采用匯聚型合成路線,根據ICH Q11的相關要求,則每個分支均應包含一個或多個起始原料。
如圖 3所示,全國原料藥工藝匯編公開了可的松醋酸酯(Ⅳ,又名醋酸可的松)的制備路線[18]。資料顯示,薯蕷皂素(Ⅰ)經三步化學合成得雙烯醇酮醋酸酯(Ⅱ),其再經八步化學合成得可的松醋酸酯。經分析,可的松醋酸酯分子中含有六個手性碳原子,其中8、9、10、13、14位手性碳的構型均來源于薯蕷皂素分子,且后續化學反應不涉及上述手性中心,同時考慮到其均為甾體母核的橋聯碳原子,因為螺環分子的剛性結構,難以發生消旋化,除反應涉及橋聯碳原子外,一般認為上述手性構型較為穩定。但是,相比薯蕷皂素,雙烯醇酮醋酸酯在雙氧水/氫氧化鈉的作用下,立體選擇性地生成α構型的環氧化物中間體(Ⅲ),其經后續化學轉化,新引入了可的松醋酸酯分子中17位的手性碳構型。
圖 3 可的松醋酸酯案例分析-異構體雜質
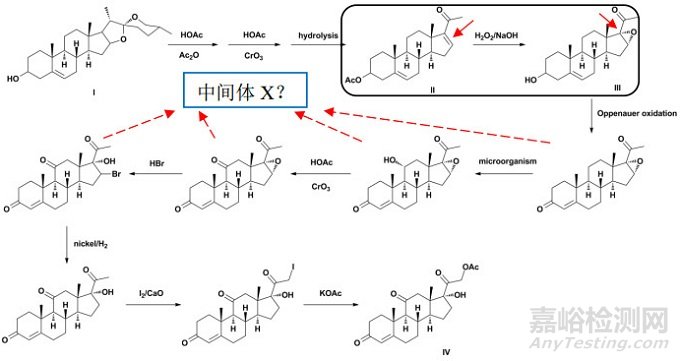
筆者認為,若不能證明中間體II經環氧化反應,立體專一地生成α構型的環氧化物,則應對潛在的β構型異構體進行研究與控制,但和ICH Q11中例4所述的事項一致,并非一定要選擇中間體Ⅱ作為合成可的松醋酸酯的起始原料。若申請人能提供相應的支持性研究數據,證明:(1)中間體Ⅱ至中間體Ⅲ中所生成的β構型異構體為“持續存在”的雜質,其構型在后續的化學轉化中是穩定的,或為定量轉化,而非部分消旋化。(2)中間體III至可的松醋酸酯中的某個已分離的中間體(Ⅹ)中,可以建立適當的(必要時,應進行規范驗證)分析方法對β構型異構體(或其衍生物)進行檢測,擬定了可接受的內控標準,提供了制訂依據,并作為原料藥質量控制策略的一部分。(3)中間體(Ⅹ)之前的工藝步驟中沒有對可的松醋酸酯雜質譜(致突變性雜質譜)有影響的雜質,也沒有經評估認為難以控制的工藝步驟。(4)同時應保證在GMP條件下有足夠的反應步驟。則可能會接受將上述工藝中稍下游的某個中間體(Ⅹ)作為起始原料,但所擬定的起始原料也應符合ICH Q11的其他相關要求。
2.3 市售化學品
ICH Q11明確若選擇市售化學品作為起始原料,考慮到其已在非藥用領域有了較為廣泛的應用,則不必論述起始原料的選擇依據。同時,ICH Q11問答文件還進一步明確了市售化學品和定制化學品的區別,不能僅依據已有穩定的市場供應(比如多個來源),就據此判斷為市售化學品。例如,本研究圖 1所示的1,3-二酮(Ⅱ)中間體,可能已有多個供應商,但基于現有認知,其僅能應用于醫藥領域,且對于制備工藝中相關雜質的來源、衍化及去除尚不十分了解,故認為其不為市售化學品。又例如,水楊酸是一種重要的有機合成原料,本身可作為原料藥用來制備多種外用皮科制劑,還可以用來合成對氨基水楊酸(4-Aminosalicylic Acid)、乙酰水楊酸(Aspirin)等藥用分子,同時也已經廣泛應用于農藥、橡膠、染料、食品及香料工業。如若選擇水楊酸作為擬定的起始原料,一般不必進行選擇依據的論述。
需要指出的是,雖然選擇市售化學品作為起始原料可被接受,但并不意味著就可以減免相關的質量研究。例如,硝呋太爾(Nifuratel)為硝基呋喃衍生物,為抗原蟲、抗真菌及抑制真菌生長繁殖的抗真菌藥,主要用于治療陰道炎及泌尿系統感染。如圖 4所示,根據Stuart Warren等提出的反向合成理論(Retrosynthetic Analysis, the Disconnection Approach)[19],硝呋太爾(Ⅰ)分子的一個潛在的合成子(Synthon)為中間體Ⅱ,并可進一步逆向分解為環氧氯丙烷(Ⅲ)。眾所周知,環氧氯丙烷是一種重要的有機化工原料和精細化工產品,主要應用于制備環氧樹脂。若申請人基于環氧氯丙烷為市售化學品,擬以其作為起始原料用于合成硝呋太爾,可能會被接受。但是,已有文獻表明[20],如圖 5所示,環氧氯丙烷主要有兩條制備路線:氯丙烯途徑和甘油途徑。其既可能作為石化產業下游產品,由氯丙烯經兩步化學合成制備,也可能作為生物柴油產業下游產品,由甘油經兩步化學合成制備。考慮到環氧氯丙烷存在不同的制備路線,反應體系(取代反應)較為復雜,且對于反應的區域選擇性尚不了解,同時部分潛在雜質為含有警示結構的潛在致突變性雜質(如氯丙烯等),若申請人不基于環氧氯丙烷生產商所提供的生產工藝,對于各實際/潛在存在雜質進行相應的研究與控制,則認為難以科學合理地制訂原料藥整體的控制策略。
圖 4 硝呋太爾反向合成分析
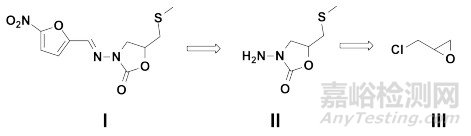
圖 5 硝呋太爾案例分析-市售化學品

3 討論
對于特定的原料藥,監管機構從未規定不同申請人應采用“相近”的合成工藝,也未要求應選擇“相同”的起始原料。起始原料的選擇應是一個具體問題具體分析(Case by Case)的過程,應根據起始原料供應商所提供的制備路線,基于申請人對于生產工藝的理解,厘清工藝的關鍵步驟及工藝參數,在全面雜質研究的基礎上,制訂相應的控制策略,從而保證原料藥的質量。
3.1 存在問題
筆者在日常審評工作中發現,部分國內注冊申報資料存在如下問題:(1)在原料藥首次注冊申報時,擬定的工藝路線僅包括一步化學合成步驟;或在原料藥批準后的生產工藝變更申請中,擬縮短原工藝路線;但均未進一步論述擬定起始原料的選擇依據;(2)未結合起始原料的合成路線,對起始原料中實際/潛在存在雜質進行相關的質量研究;(3)未明確起始原料質量標準的擬定依據;(4)未提供針對起始原料供應商詳細的審計報告,未明確是否同供應商簽訂質量保證協議和變更管理協議。上述問題反映出起始原料的選擇及其質控尚未引起足夠的重視,部分申請人尚未充分認識到起始原料在保證原料藥的質量中所起的重要作用。
另外,起始原料的選擇若不被監管機構所接受,將可能嚴重影響原料藥的審評進程。若最終重新定義起始原料,將擬定工藝前移,原擬定起始原料作為更新后的工藝路線的中間體,則需要重新提供起始原料的相關信息,包括但不限于制備路線(如,詳細的工藝流程圖)、質量標準及質量研究等,還需要根據對生產工藝的研究及理解,評估原制訂的原料藥控制策略,比如有關物質(有機雜質、無機雜質、殘留溶劑、元素雜質、潛在致突變性雜質等),是否需要進行相應的調整。值得關注的是,在今后的原料藥和制劑關聯申報過程中,還需要評估原料藥變更起始原料后對于關聯制劑的潛在影響。如若制劑已經進行了臨床試驗,則應評估相關差異是否會影響產品的質量,并提供支持性資料。
3.2 建議關注
筆者認為,ICH Q11及其問答文件所明確的起始原料選擇的原則是一個不可分割的整體,應綜合考慮所有的原則,若僅進行起始原料選擇的文字表述或是非判斷,而缺乏對于工藝的研究與理解,脫離整體風險評估及相應的控制策略,也是不恰當的。為合理地選擇起始原料,建議關注:首先,申請人是第一責任主體。結合總局第80號文的要求及其相關解讀[9],申請人應明確其既是藥品研發過程的主體,也是藥品整個生命周期中保證藥品質量的主體。申請人有義務和責任進行全面而深入的系統研發,并不斷進行多學科的自我評價,真正做到基于科學認知和研究結果,合理地選擇起始原料,而絕非僅依據日后的生產成本或變更難易。其次,應基于風險管理、并結合控制策略。為避免引入不可控的因素,進一步確保用藥安全,應從源頭開始全程控制藥品的質量。申請人應根據相關技術指導原則的要求,對雜質進行全面的分析和控制,明確可能對后續反應產生影響的雜質或可能潛在引入成品的雜質,并在此基礎上,采用適當的分析方法進行控制,制訂科學合理的內控標準。另外,考慮到起始原料選擇的復雜性,指導原則難以涵蓋各種實際問題。借鑒國外新藥上市時,監管機構和申請人會定期進行階段性討論。如有可能,建議申請人就起始原料選擇的合理性問題進行溝通交流。
參考文獻
[1] 丁恩峰, 高海燕. 原料藥國際注冊常見缺陷深度分析[J]. 醫藥工程設計, 2012, 33(6): 37-40.
[2] Top Ten Deficiencies New Applications for Certificates of Suitability for Chemical Purity[EB/OL].(2016-12)[2019-01-29]. https://www.edqm.eu/sites/default/files/cep_to_monographs_of_pheur_march2017.pdf.
[3] ICHQ11 Questions and Answers[EB/OL]. (2016-10-13)[2019-01-29]. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q11/Q11IWG_Step4_QA_2017_0823.pdf.
[4] 國家食品藥品監督管理總局. 2016年第80號國家食品藥品監督管理總局關于發布化學藥品新注冊分類申報資料要求(試行)的通告[S]. 2016.
[5] 王宏亮, 陳震. 化學合成原料藥起始物料國內外相關要求的比較[J]. 中國新藥雜志, 2014, 23(9): 998-1003.
[6] 杜爽, 梁毅. 化學合成原料藥申報過程中起始物料的選擇與控制[J]. 中國醫藥工業雜志, 2018, 49(8): 1172-1176.
[7] 顧文平, 徐敏. 化學合成原料藥申報中起始物料選擇的探討[J]. 科技創新與應用, 2016(17): 66.
[8] 操鋒, 馬玉楠. 化藥原料藥當前藥學審評技術要求初探[J]. 中國藥科大學學報, 2014, 45(3): 278-280.
[9] 黃曉龍. 化學仿制藥新申報資料要求簡介[J]. 中國新藥雜志, 2016, 25(18): 2103-2108.
[10] ICHQ7[EB/OL]. (2000-11-10)[2019-01-29]. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q7/Step4/Q7_Guideline.pdf.
[11] ICHQ11[EB/OL]. (2012-05-01)[2019-01-29]. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q11/Q11_Step_4.pdf.
[12] G.D. Searle &Co..Substituted Pyrazolyl Benzenesulfonamide for the Treatment of Inflammation: US5521207[P].1996-05-28.
[13] AUROBINDO PHARMA LTD, AMBATI V RAGHAVA REDDY, GARAGA SRINIVAS, MALLELA SAMBHU PRASAD SARMA, MEENAKSHISUNDERAM SIVAKUMARAN. AN IMPROVED PROCESS FOR THE PREPARATION OF CELECOXIB全球專利: WO2010095024(A2)[P]. 2010-08-26.
[14] 張邦樂, 梅其炳, 何煒, 等. 塞來昔布的合成[J]. 中國新藥雜志, 2002, 11(11): 859-861. DOI:10.3321/j.issn:1003-3734.2002.11.010
[15] EPAR-Scientificdiscussion[EB/OL].(2011-04-06)[2019-01-29]. https://www.ema.europa.eu/documents/scientific-discussion/onsenal-epar-scientific-discussion_en.pdf.
[16] AmbavaramVijayaBhaskar Reddy, NandigamVenugopal, GajulapalleMadhavi. A Selective and Sensitive LC-MS/MS Method for the Simultaneous Determination of Two Potential GenotoxicImpurities in Celecoxib[J]. Journal of Analytical Science and Technology, 2014, 5: 18. DOI:10.1186/s40543-014-0018-1
[17] ICH M7[EB/OL].(2017-03-31)[2019-01-29].https://www.ich.org.
[18] 《全國原料藥工藝匯編》編委會. 全國原料藥工藝匯編[M]. 北京: 國家醫藥管理總局, 1980: 530-535.
[19] Stuart Warren, Paul Wyatt.有機合成: 切斷法(原書第2版)[M].藥明康德新藥開發有限公司, 譯.北京: 科學出版社, 2010.
[20] Epichlorohydrin[EB/OL]. (2018-11-29)[2019-01-12]. https://en.wikipedia.org/wiki/Epichlorohydrin.
龔青, 仲宣惟, 田潔, 孫桂霞
1. 國家藥品監督管理局藥品審評中心, 北京 100022
2. 中國食品藥品檢定研究院, 北京 100050

來源:xml-data


