您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2024-11-27 15:41
摘要
藥品上市后變更是藥品全生命周期管理的重要環節之一,可能給藥品質量帶來潛在的影響。溶出曲線作為評價化學藥品普通口服固體制劑質量的重要指標,可揭示原研制劑與仿制制劑之間的質量一致性,因此越來越受到監管部門和藥品研發人員的重視。本文就化學藥品普通口服固體制劑上市后變更溶出曲線技術要求進行對比,并分析總結了藥品上市后變更溶出曲線評價的若干風險因素,以期對藥品上市許可持有人做好化學藥品普通口服固體制劑上市后變更研究有所助益。
關鍵詞
上市后變更;溶出曲線;口服固體制劑;原研制劑;仿制制劑
2015年8月9日,國務院印發了《關于改革藥品醫療器械審評審批制度的意見》(國發〔2015〕44號),將仿制藥由“仿已有國家標準的藥品”調整為“仿與原研藥品質量和療效一致的藥品”,強調了要以原研藥品作為參比制劑。2017年8月25日,原國家食品藥品監督管理總局發布《總局關于仿制藥質量和療效一致性評價工作有關事項的公告》(2017年第100號),進一步明確了原研藥品或在美國、日本、歐盟上市并獲得參比制劑地位的藥品均可作為參比制劑備案。自2015年仿制藥質量和療效一致性評價工作開展后,大量通過仿制藥一致性評價或視同通過一致性評價的產品陸續上市,并進入藥品集中帶量采購行列,其中化學藥品普通口服固體制劑占比較大,這些品種上市后往往伴隨著批量放大、生產場地變更、工藝變更等,如何確保一致性評價品種在省級層面的變更中既能滿足企業合理訴求,又能保證藥品質量不降低,是省級藥品監管部門值得思考及研究的方向。對于化學藥品普通口服固體制劑,溶出曲線可作為評價其質量的重要指標,能揭示原研制劑與仿制制劑之間的質量一致性;對于一些具有較好體內外相關性的藥物,溶出曲線的比較甚至可以替代體內生物等效(bioequivalence,BE)試驗?¹?;相比于單點取樣測定溶出度的方式,溶出曲線評價可以全面了解制劑的內在質量,因此越來越受到監管部門和藥品研發人員的重視。本文就化學藥品普通口服固體制劑注冊上市及上市后變更溶出曲線技術要求進行對比分析,并討論總結上市后變更溶出曲線評價的若干風險因素,以期對藥品上市許可持有人做好化學藥品普通口服固體制劑上市后變更研究有所助益。
1、國內外主要藥品監管機構對溶出曲線評價的一般要求
自2017年6月19日我國正式加入人用藥品技術要求國際協調理事會(The International Council for Harmonisation of Technical Requirements for Pharma?ceuticals for Human Use,ICH),我國藥品監管部門、制藥行業和研發機構開始逐步實施國際最高的技術標準和指南,推動著我國藥品研發和注冊與國際規則的接軌。對于藥品質量研究的溶出曲線評價方面,國際主要藥品監管機構均出臺了相關技術指導原則,美國FDA、歐盟EMA和國家藥品監督管理局(National Medical Products Administration,NMPA)的相關要求大致相同。對于溶出曲線非模型依賴相似因子(f2)的相關要求,本文就國際主要藥品監管機構相關情況進行了總結歸納,見表1。當批內藥物溶出量的相對標準偏差不符合非模型依賴的相似因子法的要求時,可考慮采用非模型依賴多變量置信區間法、f2?bootstrap法、模型依賴法等方法計算相似性。

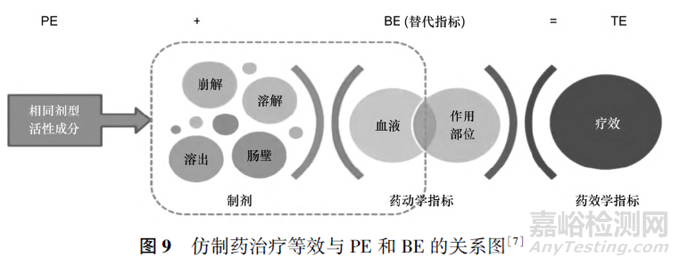
通常來說,一種仿制藥要與參比制劑雜質具有“五個相同”和“兩個等效”。“五個相同”即相同的活性成分、相同的劑型、相同的劑量、相同的給藥途徑和相同的適應證,“兩個等效”即藥學等效(pharmaceutical equivalence,PE)和BE。仿制藥只有同時滿足與參比制劑的PE和BE,才能達到與參比制劑在臨床使用上的相互替代,確保患者無論是服用仿制藥還是參比制劑均可獲得相同的療效。PE中雜質譜及溶出曲線研究是口服固體制劑的2項主要研究內容,而藥物制劑的溶出行為主要決定于藥物的溶解度、粒徑、晶型、制劑的處方與制備工藝?²?,參考《普通口服固體制劑溶出曲線測定與比較指導原則》要求,溶出行為研究推薦繪制藥物的pH?溶解度曲線,推薦選擇不少于3種pH值的溶出介質進行溶出曲線考察,如選擇pH值1.2,4.5和6.8的溶出介質。對于溶解度受pH值影響大的藥物,可能需在更多種pH值的溶出介質中進行考察。確認PE后,一般需開展BE研究,參照《以藥動學參數為終點評價指標的化學藥物仿制藥人體生物等效性研究技術指導原則》要求,對于常釋片劑和膠囊,通常采用申報的最高規格進行單次給藥的空腹及餐后BE研究。
2、上市后變更溶出曲線評價的相關要求
美國FDA、歐洲EMA和NMPA針對不同的上市后變更情形,基于風險考慮,溶出曲線研究要求存在差異,見表2。2021年2月由國家藥品監督管理局藥品審評中心(CDE)發布的《已上市化學藥品藥學變更研究技術指導原則(試行)》規定,對于制劑生產工藝的中等變更情形,要求對變更前后的樣品進行質量對比研究,變更前后樣品的溶出曲線、雜質譜、關鍵理化性質應保持一致,并符合相關指導原則的要求;對于變更制劑所用原料藥供應商情形,如變更前后制劑的溶出曲線、關鍵理化性質等存在差異,一般需考慮進行BE研究。2022年11月CDE發布《〈已上市化學藥品藥學變更研究技術指導原則(試行)〉溶出曲線研究條件的問答》,就《已上市化學藥品藥學變更研究技術指導原則(試行)》中“進行變更前后的溶出曲線對比研究”進一步說明,建議選擇變更后3批樣品與變更前樣品的代表性批次(如臨床試驗批、BE批或其他代表性批次)進行對比研究。

3、對上市后變更溶出曲線評價的若干風險因素考慮
3.1 誤差傳遞風險
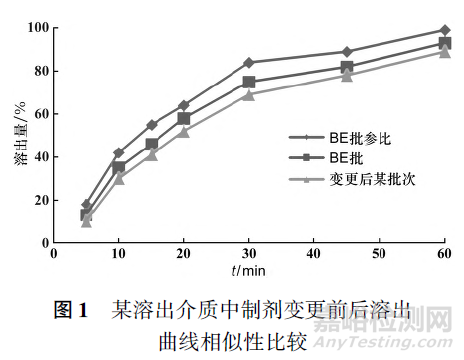
如何保證“一致性評價”不變為“一次性評價”,需要識別變更可能隱藏的內在風險。如圖1所示,制劑變更后與變更前溶出曲線雖是相似的,但可能出現變更后與變更前(BE批)相似,而與參比制劑勉強相似或不相似的情況,因此溶出曲線對比評價存在誤差傳遞風險。具體來說,如該品種在首次申報時,BE批制劑比參比制劑慢5~10個點,變更后自制制劑比BE批慢5~10個點,則變更后自制制劑與參比制劑相差值存在10~20個點的差異,導致變更后自制制劑與參比制劑體外溶出曲線不相似或勉強相似的情況。根據《〈已上市化學藥品藥學變更研究技術指導原則(試行)〉溶出曲線研究條件的問答》相關要求,與變更前樣品的代表性批次(如臨床試驗批、BE批或其他代表性批次)進行對比研究,并未要求參比制劑,但此時體內BE可能存在一定風險。如該制劑變更生產批量,關聯原料藥的供應商的變更、生產工藝變更和輔料等級變更,且為水難溶性藥物,其疊加的變更風險等級較難評估,需結合生產工藝的復雜程度、藥物特點以及變更情況等方面綜合考慮。
3.2 假陽性風險
根據由巴西國家衛生監督管理局(Brazilian Health Surveillance Agency)[3]開展的一項調研,巴西生物等效性內部數據庫(Brazilian System of Bioequivalence and Pharmaceutical Equivalence,SINEB)收集了自2008—2013年在巴西進行的所有BE研究的數據記錄,并從數據庫隨機抽樣了500個BE研究,按BCS分類進行了體內外情況統計分類,見表3和表4。巴西國家衛生監督管理局參考其分類引入了假陽性、假陰性風險,其中BCSⅡ體外溶出相似但體內BE不等效風險最高(由于BCSⅣ例數較少,并未統計在列)。


假陽性風險即體外溶出曲線區分力不足風險,即接受了不合格的產品,此種情況在注冊申報期間可能遇到。如在首次申報處方工藝開發時pH1.2,4.5和6.8溶出介質中自制制劑溶出曲線與參比制劑相似,認為已達到了PE,見圖2~圖4。



正式開展BE研究時,研究結果表明生物不等效,綜合分析生物不等效數據和藥品特性,經過探索優化后,申請人得到能區分參比制劑和自制制劑差異的溶出介質(比如pH5.5溶出介質),pH5.5溶出介質中自制制劑與參比制劑的溶出曲線相似,且不影響pH1.2,4.5和6.8溶出介質與參比制劑的相似性,體內BE符合要求,見圖5。

后續發生上市后變更,變更后上述溶出介質pH1.2,4.5和6.8溶出介質溶出情況與首次申報時相似,若申請人此時未研究有區分力的pH5.5溶出介質(該介質在首次申報時可能被定義為質量控制介質,變更時需提交該介質的溶出情況),則該變更缺失了一個重要的評估指標。上市后變更提供的溶出介質是否具備區分力,需結合《〈已上市化學藥品藥學變更研究技術指導原則(試行)〉溶出曲線研究條件的問答》相關要求及品種特性、藥動學特征綜合評判,對于一些高風險的品種、高風險的變更情況,也可以參考日本橙皮書中同品種溶出曲線的溶出介質要求及溶出條件。
3.3 假陰性風險
注冊申報上市及上市后變更溶出曲線可能存在過度區分風險,即溶出曲線區分力過度。企業通過改變制劑處方、生產工藝,得到不同體外釋放的溶出曲線,但體內釋放無明顯差異,即體外溶出不相似但體內BE,這種情況對產品質量的影響的風險遠遠小于假陽性的風險,但容易造成企業不必要的“束縛”,往往會過猶不及,可分為2種情況。
第1種情況見圖6和圖7,體外多條溶出介質溶出曲線不相似,但體內BE符合要求,這種情況申報上市可能會遇到,需結合制劑特性分析溶出曲線不相似的原因。體外溶出曲線可以用于產品質量監控,便于在上市后變更研究及質量回顧時更好地控制產品質量。開展上市后變更研究時,變更后樣品在該溶出介質中的溶出曲線需與變更前關鍵批次(如BE批)相似,可不與參比制劑相似。


第2種情況見圖8,在實際上市后變更中,變更情形不符合《〈已上市化學藥品藥學變更研究技術指導原則(試行)〉溶出曲線研究條件的問答》的意見,某制劑持有人對變更后制劑進行溶出曲線考察,與變更前驗證批比較,在水、0.1mol·L-1鹽酸中的f2>50,在pH6.8緩沖鹽(標準介質)中15min溶出度均≥85%,溶出行為相似,但在pH4.5緩沖鹽中的溶出行為不相似;與BE批比較,在水、0.1mol·L-1鹽酸、pH6.8緩沖鹽(標準介質)中溶出行為相似,在pH4.5緩沖鹽中的溶出行為不相似;與參比制劑比較,在0.1mol·L-1鹽酸、pH6.8緩沖鹽(標準介質)中溶出行為相似,在水、pH4.5緩沖鹽中的溶出行為不相似。根據原研歐洲EMA審評報告的結論,該制劑在pH4.5左右對pH的變化非常敏感,采用pH4.5溶出介質的溶出方法存在過度區分力的問題,可能會出現f2值偏低的情況。基于歐洲EMA審評報告,結合藥物活性物質(active pharmaceutical ingredient,API)的特性,認為變更后制劑在pH4.5溶出介質溶出曲線與變更前不相似的情況對產品質量影響可能較小。

3.4 特殊品種風險
根據《已上市化學藥品藥學變更研究技術指導原則(試行)》,對于治療窗窄的藥物或水難溶性藥物的普通口服固體制劑,涉及重大變更對藥品安全性、有效性和質量可控性均可能產生較顯著的影響,一般需考慮進行BE研究。變更指導原則中特別強調了“治療窗窄的藥物或水難溶性藥物的普通口服固體制劑”,在變更研究中應特別予以關注。如對于卡馬西平片,《日本溶出曲線數據庫》擬定5min不得超過60%和30min不得少于70%;美國藥典擬定10min釋放量應為30%~50%,45min時不得少于75%。兩點法的規定既限定了具體溶出行為,又可有效防止處方中加入大量表面活性劑或增溶劑的做法,值得借鑒[5]。巴西國家衛生監督管理局統計數據見表5,BCSⅡ類體內BE不等效的風險是水易溶性藥物的2.5~4倍?³?。另外,美國FDA指南提到,若藥品上市后變更(如組分、成分及生產工藝變更)符合指南中BCSⅠ類或BCSⅢ類相關要求且變更前后產品的溶出情況相似,可以考慮豁免BE研究???,預示著水難溶性藥物可能存在較高的風險及監管要求。
4、分析與討論
通過PE+BE可綜合判定產品是否可通過質量和療效一致性評價,見圖9。上市后中等變更情形雖然在設定的范圍內變動,但僅通過藥學評價也可能存在一定風險。如反應吸收程度的指標AUC的等效判定范圍為80%~125%,假設變更前企業產品的吸收量與參比制劑比較為85%,這種情況可得到二者BE的結果;若變更后產品與變更前產品比較溶出相似,相似因子為50(勉強相似,若做BE研究與變更前吸收量可能為85%),也可得到二者BE的結果。但相對于原研產品來說,變更后體內存在誤差傳遞效應,變更后產品的吸收量可能僅為72%,顯然不能認為二者BE,上述風險是客觀存在的。《〈已上市化學藥品藥學變更研究技術指導原則(試行)〉溶出曲線研究條件的問答》建議,選擇變更后3批樣品與變更前樣品的代表性批次(如臨床試驗批、BE批或其他代表性批次)進行對比研究,通過一致性評價品種BE批是變更前典型代表性批次。而《以藥動學參數為終點評價指標的化學藥物仿制藥人體生物等效性研究技術指導原則》要求,BE研究時應與參比制劑比較。上述指導原則中比較目標的不一致,可能會引起不同程度的誤差傳遞效應,因此應關注參比制劑、自制制劑的體外溶出差異及體內吸收差異之間的關系。
藥品注冊申報上市審評時主要依據BE研究+體外多溶出介質溶出曲線獲批上市,BE研究為“金標準”,體外多溶出介質曲線可以不相似;而上市后變更的中等變更備案事項,一般僅要求提供多溶出介質溶出曲線對比,因無BE研究的“金標準”,因此多溶出介質溶出曲線的“區分力”則變得尤為重要。但是,溶出曲線的區分力并沒有統一的概念和標準,故《〈已上市化學藥品藥學變更研究技術指導原則(試行)〉溶出曲線研究條件的問答》指出,“某些標準介質中可能含有少量表面活性劑,除標準介質外,其余溶出介質通常不建議添加表面活性劑,需提供不加表面活性劑的溶出曲線研究數據,并進行相似性評估”、“除標準介質外,其余溶出介質通常不建議調整轉速”,可能是避免因高轉速、表面活性劑的添加等此類降低區分力的方法而導致變更前后的偽相似。
溶出曲線研究的目的在于評估變更前后樣品之間的相似性,通過相似性評估關鍵質量屬性的一致性,進而可能影響到變更類別評判,如溶出曲線“不相似”,變更級別可能由“中等”上升為“重大”。但溶出曲線相似不一定“萬事大吉”,如f2由變更前與BE批相比的“70”變化為變更后與BE批相比的“51”,可能提示工藝過程控制或物料屬性的變化對溶出行為產生了不利影響,需結合變更情況綜合考量。
4.1 相關建議
上市后變更溶出曲線相似比較首先應參照《〈已上市化學藥品藥學變更研究技術指導原則(試行)〉溶出曲線研究條件的問答》相關要求進行研究,同時關注CDE官網2023年12月14日發布的“化學仿制藥共性問題28則有關溶出曲線的問題回答”。上述指導原則出臺前已經批準了大量的一致性評價品種,同時指導原則的實施往往存在一定的過渡期,在此期間,若首次申報質量對比中多介質溶出曲線的介質、方法與上述不同,建議企業在變更時提交首次注冊申報時相關溶出介質的溶出數據及方法,并與變更后樣品進行對比。
《〈已上市化學藥品藥學變更研究技術指導原則(試行)〉溶出曲線研究條件的問答》建議選擇變更后3批樣品與變更前樣品的代表性批次(如臨床試驗批、BE批或其他代表性批次)進行對比研究,如為仿制制劑,強化了BE批次的重要性,并未要求與參比制劑對比,基于一致評價時參比制劑的重要地位,對某些變更情形,同時考慮到誤差傳遞的風險,建議企業提供參比制劑的多介質溶出曲線情況,如基于BCS分類豁免的品種變更,變更后產品需要和參比制劑進行對比,處方和溶出曲線仍需要符合ICH M9的要求。
基于不同BCS分類藥物溶出曲線相似及體內BE等效的難易程度不同,變更時可以考慮藥物的BCS分類情況,并結合藥物制劑的體內吸收特點,對變更情況進行全面的評估。
特殊品種上市后變更可能需特別關注,如治療窗窄的藥物、濃度依賴型抗生素,其體內療效及不良反應與血藥濃度高度相關的,建議綜合參考國內外相關溶出曲線研究技術要求。
對于一些高風險變更情況,參照《〈已上市化學藥品藥學變更研究技術指導原則(試行)〉溶出曲線研究條件的問答》相關要求進行研究外,也可以結合日本橙皮書中同品種溶出曲線的溶出情況進行研究。
4.2 展望
上市后品種尤其是通過一致性評價品種,往往積累了一定的藥學研究數據及體內BE研究數據,如何結合品種變更情況利用這些數據,實現更精準地把控溶出曲線風險,而不是機械簡單地判斷“相似”或“不相似”值得思考,如某通過一致性評價的口服固體制劑,進行了體內BE研究,通過體內BE試驗結果結合API及制劑相關特性之后反推與體外溶出情況的關聯性,或者引入模型評價預測手段???¹²?,進行數據挖掘利用,利用模型預測藥物體內行為,輔助變更前后藥物溶出差異的評估,建立有預測力(與BE相關或體內吸收情況相關)的溶出曲線評價方法,有時可以輔助排除“假陽性”或“假陰性”風險,從而提高上市后變更溶出風險把控。
來源《中國新藥雜志》 2024年 第33卷第21期

來源:凡默谷


