您當(dāng)前的位置:檢測(cè)資訊 > 實(shí)驗(yàn)管理
嘉峪檢測(cè)網(wǎng) 2024-11-15 08:39
概念及原理
無縫克隆可將插入片段定向克隆至任意載體的任意位點(diǎn),不需要任何限制性內(nèi)切酶和連接酶,重組效率高。將載體進(jìn)行線性化,在插入片段正/反向PCR引物5’端引入線性化載體的末端序列,使得PCR產(chǎn)物5’和3’最末端分別帶有和線性化載體兩末端一致的序列(15 - 20 bp)。這種PCR產(chǎn)物和線性化載體按一定比例混合后,在重組酶的催化下,37℃反應(yīng)30 min即可進(jìn)行轉(zhuǎn)化,完成定向克隆。
過程圖
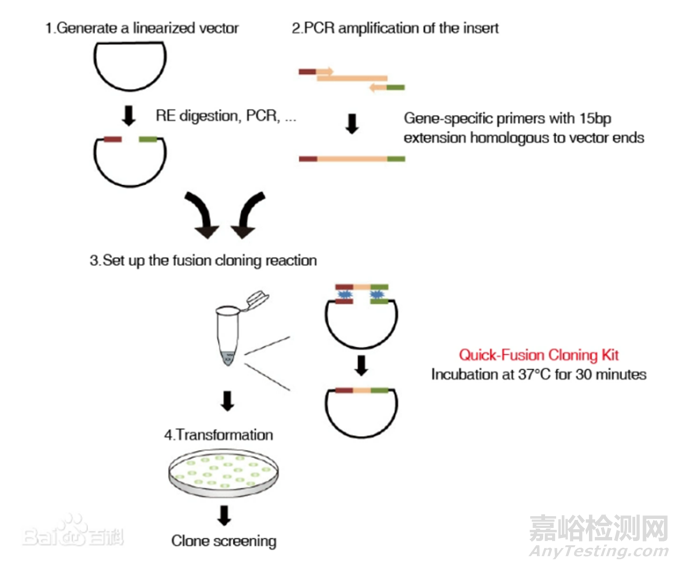
線性化載體的制備
選擇合適的克隆位點(diǎn),對(duì)載體進(jìn)行線性化。盡量選擇無重復(fù)序列且載體克隆位點(diǎn)上下游20 bp區(qū)域內(nèi)GC含量在40% - 60%之間的位點(diǎn)進(jìn)行克隆。線性化的方法有酶切制備和反向PCR擴(kuò)增制備。
(1)酶切制備
酶切制備線性化載體時(shí),推薦使用雙酶切方法使載體線性化完全,降低轉(zhuǎn)化背景(假陽(yáng)性克隆);若使用單酶切線性化,請(qǐng)適當(dāng)延長(zhǎng)酶切時(shí)間以減少環(huán)狀質(zhì)粒殘留,降低轉(zhuǎn)化背景。
注:a. 經(jīng)雙酶切進(jìn)行線性化的載體無需去磷酸化。反應(yīng)體系內(nèi)無DNA連接酶,不會(huì)引發(fā)載體自連。因此,即使是單酶切方式制備的線性化載體也無需進(jìn)行末端脫磷酸處理;b. 酶切完成后,應(yīng)快速將內(nèi)切酶失活或?qū)δ康漠a(chǎn)物純化后再用于重組反應(yīng);c. 酶切后進(jìn)行膠回收純化,純化后通過瓊脂糖凝膠電泳檢測(cè)產(chǎn)物。d. 重組產(chǎn)物轉(zhuǎn)化后出現(xiàn)的假陽(yáng)性克隆(無插入片段),是由未線性化環(huán)狀載體轉(zhuǎn)化而形成。
(2)反向PCR擴(kuò)增制備
推薦使用高保真聚合酶擴(kuò)增減少堿基突變的概率。50μl的PCR體系中,推薦使用0.1 - 1ng環(huán)狀質(zhì)粒模板,或使用預(yù)線性化質(zhì)粒作為模板,以減少環(huán)狀質(zhì)粒模板殘留對(duì)克隆陽(yáng)性率的影響。當(dāng)模板為環(huán)狀質(zhì)粒時(shí),擴(kuò)增產(chǎn)物建議用Dpn I(甲基模板消化酶)消化后使用。
注:通過膠回收純化PCR產(chǎn)物,推薦純化后通過瓊脂糖凝膠電泳檢測(cè)其質(zhì)量和濃度。
插入片段的制備
(1)引物設(shè)計(jì)總原則
插入片段擴(kuò)增引物由兩部分構(gòu)成:重疊區(qū)域 + 特異性引物。在插入片段正反向擴(kuò)增引物的5’端引入線性化載體兩末端同源序列,使擴(kuò)增后的插入片段5’和3’最末端分別帶有和線性化克隆載體兩末端對(duì)應(yīng)一致的同源序列(15 - 20 bp,不包括酶切位點(diǎn))。
插入片段正向擴(kuò)增引物設(shè)計(jì)方式為:
5’-上游載體末端同源序列 + 酶切位點(diǎn)(可保留或刪除)+ 基因特異性正向擴(kuò)增引物序列-3’
插入片段反向擴(kuò)增引物設(shè)計(jì)方式為:
5’-下游載體末端同源序列 + 酶切位點(diǎn)(可保留或刪除)+ 基因特異性反向擴(kuò)增引物序列-3’
▲基因特異性正/反向擴(kuò)增引物序列即常規(guī)插入片段正/反向擴(kuò)增引物序列,Tm值60 - 65℃為佳;▲上/下游載體末端同源序列即線性化載體最末端序列(用于同源重組),GC含量40% - 60%為佳;▲計(jì)算擴(kuò)增引物Tm值時(shí),只需計(jì)算特異性引物的Tm值,引入的額外序列無需計(jì)算。
插入片段PCR擴(kuò)增
插入片段可用任意PCR酶(Taq酶或高保真酶)擴(kuò)增,無需考慮產(chǎn)物末端有無A尾(重組過程中將被去除,在最終載體中不會(huì)出現(xiàn))。但為了減少擴(kuò)增突變的引入,推薦使用高保真聚合酶。
線性化載體與插入片段的使用量
最適克隆載體使用量 = [0.02 × 克隆載體堿基對(duì)數(shù)] ng (0.03 pmol)
最適插入片段使用量 = [0.04 × 插入片段堿基對(duì)數(shù)] ng (0.06 pmol)
注:a.插入片段長(zhǎng)度大于克隆載體時(shí),最適克隆載體與插入片段使用量的計(jì)算方式應(yīng)互換,即將插入片段當(dāng)做克隆載體,克隆載體當(dāng)做插入片段進(jìn)行計(jì)算。b.線性化克隆載體的使用量應(yīng)在50 - 200ng之間;插入片段擴(kuò)增產(chǎn)物的使用量應(yīng)在10 - 200ng之間。當(dāng)使用上述公式計(jì)算DNA最適使用量超出這個(gè)范圍時(shí),直接選擇最低/最高使用量即可。c.線性化克隆載體和插入片段擴(kuò)增產(chǎn)物未進(jìn)行DNA純化直接使用時(shí),加入總體積應(yīng)不超過反應(yīng)體系體積的1/5,即4μl。
重組反應(yīng)
按照說明書配置體系反應(yīng)
無縫克隆實(shí)驗(yàn)常見問題與解決辦法
(1)目的片段大,不好克隆或克隆容易有突變?cè)趺崔k?
PCR進(jìn)行目的片段擴(kuò)增時(shí)一定要采用高保真酶,對(duì)于大片段的擴(kuò)增要采用兼容大片段的高保真酶,推薦TAKARA的高保真酶,可適用于10K以內(nèi)的片段擴(kuò)增。
(2)目的片段擴(kuò)增時(shí)出現(xiàn)非特異性條帶
1.PCR引物設(shè)計(jì)的問題,可設(shè)計(jì)多對(duì)引物,選擇出特異性最好的一對(duì)。
引物設(shè)計(jì)原則:
1)引物長(zhǎng)度常用的是18-27bp,但不應(yīng)大于38bp,因?yàn)檫^長(zhǎng)會(huì)導(dǎo)致其延伸溫度太高,不適合 DNA 聚合酶進(jìn)行反應(yīng)
2)3’端最好不要有A,5’端可以容忍二聚體,但3’端一定不要有二聚體
3)不要有連續(xù)的GGG 或CCC、GCGC、CGCG之類,不要有錯(cuò)配,前后引物的退火溫度要差不多,差異在2度以內(nèi)
4)避免引物自身或與引物之間形成4個(gè)或4個(gè)以上連續(xù)配對(duì),避免引物自身形成環(huán)狀發(fā)卡結(jié)構(gòu)
5)Tm值在55-65℃,GC含量在40%-60%
6)引物之間的Tm值相差避免超過2℃
7)引物的3’端避免出現(xiàn)3個(gè)或3個(gè)以上連續(xù)相同的堿基,引物3'端要避開密碼子的第3位,因密碼子的第3位易發(fā)生簡(jiǎn)并,會(huì)影響擴(kuò)增的特異性與效率
8)引物的設(shè)計(jì)好后使用Blast 驗(yàn)證
2.優(yōu)化PCR反應(yīng)體系,可考慮優(yōu)化退火溫度等PCR參數(shù)。

來源:實(shí)驗(yàn)老司機(jī)


