有關物質檢查研究的主要內容包括分析方法的建立與驗證����、雜質限度的確定。本文將針對這兩方面的內容,討論有關物質的研究要求,并指出申報資料中需要注意的幾個問題�。
1����、分析方法的建立與驗證
有關物質研究的首要問題是建立合適的分離和檢測方法�。有關物質的檢查方法包括化學法、光譜法和色譜法等,其中色譜法最為常用。通常��,可基于原料藥的合成工藝初步分析藥品中可能存在的工藝雜質����,同時基于主藥的結構分析其可能的降解途徑及降解產物���,在此基礎上根據主藥及雜質的理化性質��、化學結構及雜質的控制要求等考慮選用合適的檢查方法���,例如HPLC����、TLC���、GC 或CE 等。
分析方法的建立包括分離檢測條件的篩選和優化、方法的驗證兩個方面的研究工作��。以有關物質檢查最為常用的HPLC-UV 方法為例���,在方法的篩選和優化研究中需要完成三部分的工作���,即確定分離和檢測條件���、確定待測樣品的制備方法以及雜質的定量方法�����。其中�,分離和檢測條件關系檢查方法的專屬性和靈敏度�����,是方法建立過程中最為重要也最為復雜的研究內容�����。一般而言����,分離和檢測條件的確定包括以下幾個步驟:
⑴明確方法的檢測目標并盡可能了解待測物質的理化性質���,選擇合適的含有潛在工藝雜質或降解產物的預測性樣品(predictive sample)。例如原料藥粗品�����、重結晶母液�����、破壞試驗樣品及已分離出的雜質等用于色譜條件的篩選�。
?��、坪Y選固定相和流動相條件�����。通常是基于文獻和經驗����、待測物質的理化性質�,以及研究者對色譜柱、溶劑和緩沖液等性質的了解����,選擇初步的色譜柱����、流動相����,并采用選定的預測性樣品對分離條件進行考察����。重復這一篩選過程直至找到一個可以使各主要雜質及主藥之間分離情況達到基本可接受水平的分離條件作為初始色譜條件。
?���、菍Τ跏忌V條件進行優化��。設計一系列試驗考察色譜參數,如有機相比例����、pH����、流速、柱溫和檢測波長等對雜質分離和檢出的影響�,并根據試驗結果確定最優色譜參數�。在優化研究過程中還可以觀察到哪些參數對方法有顯著影響���,對這些參數應在方法驗證研究的耐用性試驗中予以考察����。
一些仿制藥品的有關物質檢查方法在國內外藥典中有收載或已有文獻報道,上述過程可能會簡化���。但是,在無文獻可以參考時�����,開發一個新方法的工作量會比較大��,花費的時間也比較長。近年來����,在有關物質檢查方法的開發研究中應用了一些高效的方法開發策略����,特別是計算機輔助的HPLC 色譜條件篩選方法發展很快���,提高了篩選的效率�����。
在確定檢查的色譜條件�、分析樣品的制備方法以及雜質的定量方法后�����,應按照相關技術指導原則的要求開展方法學驗證工作( 中國藥典2010年版二部附錄172)�����。需要注意的是,方法驗證工作是在檢查方法確定后開展的,其目的是證明采用的方法適用于相應檢測要求,檢查方法的篩選優化研究不能代替方法的驗證工作,盡管二者的部分研究內容有交叉。
在方法的建立和驗證過程中��,尚需要關注以下幾個問題:
?、挪捎肏PLC-UV 法進行檢查時,使用二極管陣列檢測器有著重要意義,通過該檢測器可獲得各雜質的紫外光譜信息��,有助于判斷雜質檢測波長選擇的合理性�����,避免雜質的漏檢,同時還可以進行峰純度檢查���。
⑵優化的檢測波長應是主要雜質的最大吸收���,而不是主藥的最大吸收,同時���,應使主藥和各主要雜質的響應差異最小。如果主藥和各主要雜質的響應差異較大�,例如超出了0.9 ~ 1.1 的范圍時����,在采用主成分自身對照法進行雜質定量時需要加校正因子��。此外��,如用UV 低波長檢測時,還應考慮有機相中有機溶劑的截止使用波長����。
?�、沁m度的破壞試驗可用于考察分析方法對降解產物的檢出能力,也可用于考察主藥的降解途徑和降解產物�����。但是,破壞試驗的目的不是使活性化合物完全降解���,而只是要產生少量的降解產物,通常在敏感條件下活性化合物降解10%~ 30%較為合適,過分降解將產生一些在生產和貯存條件下不可能生成的二級甚至三級降解產物�,對色譜條件的篩選不利���。在破壞試驗中應關注物料平衡情況����,但是由于降解產物和主藥的響應可能存在差異或降解產物無法被檢出�、降解產物可能進一步降解以及存在多條降解途徑等因素���,物料不平衡的現象還是較為常見�����。
?����、仍诜椒炞C的耐用性試驗中應對色譜系統的適用性進行考察,并通過耐用性評估�����,建立一系列的系統適用性參數( 例如分離度等)���,以確保在任何時候使用該分析方法都是有效的��。和方法優化研究中對色譜參數的考察不同��,耐用性試驗中各試驗參數應在一個比較窄的范圍內變化以模擬正常的日間變異。
2�����、雜質限度的確定
雜質控制的主要目的是將有害雜質控制在安全范圍內以保證臨床用藥的安全性�,次要目的是合理控制藥品批間和有效期內雜質水平的波動以保證藥品質量的均一性。因此����,安全性決定藥品中所能接受的雜質水平,是確定雜質限度的首要考慮因素����。如果某雜質是主藥在動物或人體中的主要代謝產物�����,則可不考慮其安全性。另外,對于具有基因毒性和致癌性的雜質,在確定限度時應予以特別關注���。在符合安全性要求的前提下,確定雜質限度還要結合工藝的可行性和貯存的方便性考慮。
獲取雜質安全性信息主要有3 種途徑。第一�,針對某雜質進行安全性研究直接獲取安全性信息���,研究用樣品可以采用含有較高水平該雜質的原料藥或制劑�����,也可以采用分離出的雜質單體;第二,通過和已上市產品進行雜質對比研究間接獲取雜質的安全性信息;第三,通過檢索文獻獲取某特定雜質的毒性數據。
對于大多數創新藥�����,雜質安全性信息主要來源于整個研發過程中各項安全性研究結果�,包括非臨床毒理研究和臨床研究。一般而言�����,在創新藥開發過程中�����,按照毒理研究樣品�����、臨床研究樣品和上市產品的順序,其中的雜質含量應依次遞減,毒理研究用樣品中的雜質含量應該最高���。上市產品雜質限度的確定要基于對整個研發過程中雜質數據的積累和分析,特別是臨床前主要安全性研究、臨床研究所用樣品的雜質數據���,以及規模化生產條件下產品的雜質數據。例如,某一創新藥的日給藥量為0.5 g�����,原料藥雜質數據和擬定限度見表1�����。

基于表1 中的雜質數據對原料藥擬定雜質限度的合理性分析如下:
對于雜質A�����,0.2%的擬定限度高于毒理和臨床研究用批次的檢測結果,也高于規模化生產批次的實測結果,限度依據不充分;按照雜質研究相關技術指導原則的要求,同時考慮到規模化生產批次雜質數據,降低到0.1%較為合理���。
對于雜質B,0.2%的限度有毒理/ 臨床研究批次的雜質數據支持,但和規?;a批次的實測結果相比過緊;放寬到0.3%仍可以保證安全性�,同時也可避免實際生產的雜質B 含量在0.2%~ 0.3%之間的產品被判為“ 不合格”���。
對于雜質C����,0.2%的限度有毒理/ 臨床研究批次的雜質數據支持,也和規模化生產批次的實測結果相當,因此可認為是合理的�。
對于雜質D��,0.1%的限度高于毒理/ 臨床研究批次�����、規?�;a批次的檢測結果,但符合相關指導原則的要求�,是可以接受的����。
單個未知雜質0.1%的限度符合相關指導原則的規定�����。未知雜質總量(0.5% ) 和雜質總量(1.0% ) 分別與各種批次的雜質數據��、單個雜質限度之和吻合,均是可以接受的��。
對于仿制藥( 包括仿制國內��、外上市藥品)��,研制產品與已上市原研產品的雜質對比研究是論證雜質安全性和限度的較為簡便的方法。另外�,對于特定雜質的限度也可引用藥典標準���、文獻進行論證��。
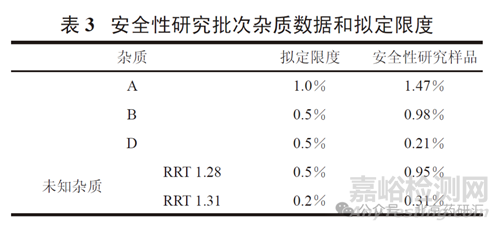
但是,通過上述途徑無法論證時���,例如研制產品中含有已上市參照品中未含有的新雜質,且含量超過了相關技術指導原則規定的質控限度���,則需要進行安全性研究以獲取雜質的直接安全性數據。例如���,某一仿制原料藥( 最大日劑量為300 mg) 的雜質數據見表2����。
表2 數據顯示����,仿制品和上市參照品的雜質譜不完全相同�,包括雜質種類和主要雜質含量均有較大差別。仿制品中三個已知雜質(A�、B��、D) 的含量均高于上市品,且高于ICH 指導原則規定的界定閾值(qualifi cation threshold)0.15% [2]�����。其中�����,雜質B 本身為一上市藥物��,有長期人用歷史,雜質D為主藥在人體內產生的主要代謝產物����,在制定限度時可以不考慮安全性問題�����。此外,仿制品中的兩個未知雜質(RRT 1.28 和1.31) 也高于 0.15%�。且缺乏試驗和文獻資料支持其安全性��,需要進行必要的安全性研究。安全性研究(遺傳毒性試驗���、重復給藥毒性試驗) 所用仿制品批次的雜質數據見表3����。安全性研究結果支持所擬定的各雜質限度。
3、提交申報資料需要關注的問題
為幫助審評人員準確了解有關物質的研究和控制情況�����,國內注冊的申報資料中應按照指導原則及其它相關規定的要求提供全面��、詳細的有關物質研究信息���,需要關注以下問題:
?�、艑υ纤幵诤铣伞⒕坪唾A存過程中實際或可能產生的雜質進行綜述;對制劑生產和穩定性考察中觀察到的降解產物進行綜述�,包括原料藥與輔料或內包裝材料���、封閉物之間的反應產物���。綜述中應包括雜質的化學名稱����、結構式和來源分析( 例如合成起始原料��、反應中間體�、反應副產物和降解產物等)�。
⑵對于新建的有關物質檢查方法,應提供方法的研究報告�,包括試驗資料和支持性圖譜���,以清晰展示檢查方法的建立和優化過程;采用藥典���、文獻方法�,如對試驗參數進行了調整和優化���,亦應提供調整前后的對比研究資料����。同時�����,應根據分析方法的目的����,提供完整的驗證資料,注意根據耐用性試驗結果建立合適的系統適用性要求。
⑶對于創新藥,需要提供藥品研制過程中所有批次樣品( 包括用于安全性����、臨床研究的樣品) 的雜質檢測數據�,并對整個研發過程中的不同規模樣品的雜質情況進行比較��,對差異進行討論�����。對于仿制藥以及已上市產品的改劑型產品,一般還要提供和已上市產品的有關物質對比檢測數據。基于上述數據以及穩定性研究中雜質檢查數據��,詳細論述各雜質限度確定的依據�。
⑷對于藥品中實際存在的、含量大于鑒定限度的雜質應說明結構研究情況����,已鑒定結構的應提供結構研究資料���,未能鑒定結構的亦應說明所開展的研究��。如研究或質量標準中使用了雜質對照品�,要提供雜質對照品的來源或制備工藝、結構確證�����、純度標定和質量標準等資料����。
⑸按照藥品審評中心發布的《藥品研究色譜數據工作站及色譜數據管理要求( 一)》附件1 的要求提供規范的支持性圖譜�,包括方法研究和驗證的全部圖譜��、穩定性研究中的全部圖譜及代表性批次的圖譜。
4�����、結語
有關物質是影響藥品安全性的因素之一�����,而有關物質檢查研究是目前我國藥品研發中的薄弱點之一�����。要全面提升我國藥品研發的水平、切實保證公眾用藥的安全性���,必須重視并加強有關物質的研究。