藥品研發(fā)實(shí)驗(yàn)記錄是指在藥品研發(fā)過程中�,應(yīng)用實(shí)驗(yàn)、觀察���、調(diào)查或資料分析等方法��,根據(jù)實(shí)驗(yàn)實(shí)際情況直接記錄或統(tǒng)計(jì)形成的各種數(shù)據(jù)�、文字、圖表、照片等原始資料��。應(yīng)規(guī)范管理藥品研發(fā)實(shí)驗(yàn)記錄��,保證研發(fā)實(shí)驗(yàn)記錄�����、數(shù)據(jù)的真實(shí)、準(zhǔn)確��、完整����、可追溯。
眾所周知���,只有通過官方的技術(shù)審查、研制和生產(chǎn)現(xiàn)場(chǎng)核查�,產(chǎn)品才能獲得注冊(cè)批準(zhǔn)����,國(guó)家藥品監(jiān)督管理局食品藥品審核查驗(yàn)中心在2021年12月20日發(fā)布了《藥品注冊(cè)核查要點(diǎn)與判定原則(藥學(xué)研制和生產(chǎn)現(xiàn)場(chǎng))(試行)》�,自2022年1月1日起施行;其中藥學(xué)研制現(xiàn)場(chǎng)核查是通過對(duì)研究過程中原始記錄、數(shù)據(jù)及現(xiàn)場(chǎng)進(jìn)行核實(shí)和/或?qū)嵉卮_認(rèn),核實(shí)相關(guān)申報(bào)資料的真實(shí)性�����、一致性����。
而藥學(xué)研發(fā)實(shí)驗(yàn)記錄是撰寫藥品申報(bào)資料的依據(jù),申報(bào)資料中總結(jié)或提煉的實(shí)驗(yàn)所使用的物料、儀器設(shè)備���,采用的實(shí)驗(yàn)條件����、實(shí)驗(yàn)方法、操作步驟��、實(shí)驗(yàn)過程���,觀察到的現(xiàn)象���,測(cè)定的數(shù)據(jù)�,得出的結(jié)果結(jié)論等均在藥學(xué)研發(fā)實(shí)驗(yàn)記錄中有記載和體現(xiàn)。只有規(guī)范管理藥學(xué)研發(fā)實(shí)驗(yàn)記錄,保證研發(fā)實(shí)驗(yàn)記錄�、數(shù)據(jù)的真實(shí)����、準(zhǔn)確���、完整����、可追溯,才能通過藥學(xué)研制現(xiàn)場(chǎng)核查���,確保產(chǎn)品獲得注冊(cè)批準(zhǔn)。
藥學(xué)研發(fā)實(shí)驗(yàn)記錄的管理包括設(shè)計(jì)��、書寫����、審核、保管�����、借閱等流程�����,記錄的審核是其中重要的環(huán)節(jié)和步驟,記錄審核可以及時(shí)的發(fā)現(xiàn)問題���、改進(jìn)問題,通過總結(jié)���、分析以提高記錄審核的效率���,并反過來進(jìn)一步促進(jìn)記錄的設(shè)計(jì)����、記錄的規(guī)范書寫�,本次就藥學(xué)研發(fā)實(shí)驗(yàn)記錄的審核要點(diǎn)進(jìn)行總結(jié)。
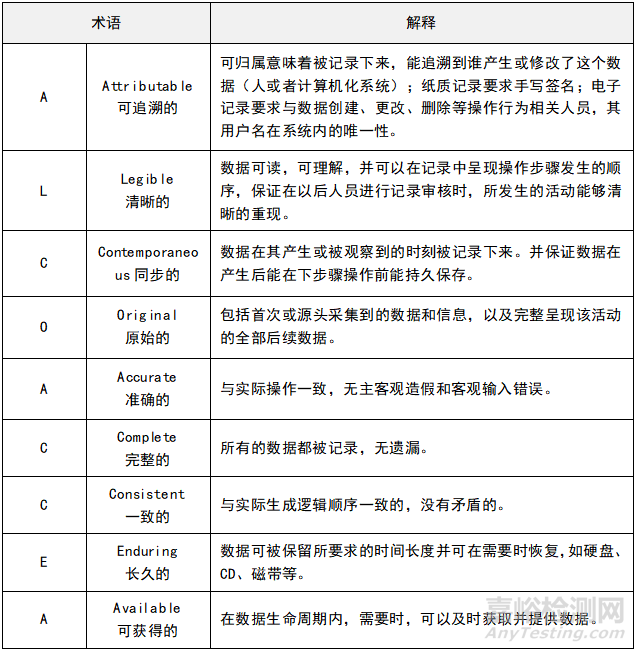
一�、要熟悉數(shù)據(jù)可靠性(ALCOA+)的原則�����,適用于藥品數(shù)據(jù)全生命周期的管理。
二��、要組織各個(gè)部門建立實(shí)驗(yàn)記錄的通用模板或?qū)倌0?��,便于記錄格式的統(tǒng)一���、書寫和審核����。實(shí)驗(yàn)記錄的內(nèi)容通常應(yīng)包括實(shí)驗(yàn)名稱、實(shí)驗(yàn)?zāi)康?��、?shí)驗(yàn)設(shè)計(jì)或方案���、實(shí)驗(yàn)時(shí)間�、實(shí)驗(yàn)材料、實(shí)驗(yàn)方法����、實(shí)驗(yàn)過程����、觀察指標(biāo)�����、實(shí)驗(yàn)結(jié)果和結(jié)果分析等內(nèi)容��,具體內(nèi)容可根據(jù)研究的內(nèi)容來確定。
三��、應(yīng)明確記錄審核的責(zé)任�,通過班組、部門�����、QA的層層審核�����,確保記錄符合數(shù)據(jù)完整性的原則�。
四��、建立通用的審核要點(diǎn)��,便于各人員、各部門進(jìn)行審核����,提高審核效率。


五、在審核、抽查記錄過程中應(yīng)注意及時(shí)總結(jié),對(duì)共性問題進(jìn)行培訓(xùn)教育�,提高人員記錄書寫的規(guī)范性�,提高對(duì)記錄的認(rèn)識(shí)�,也可提高記錄審核的效率。共性問題包括但不限于:實(shí)驗(yàn)信息不完整、記錄不及時(shí)或漏記�、修改不規(guī)范�����、實(shí)驗(yàn)人員/復(fù)核人員未簽名簽日期等,在培訓(xùn)時(shí),要結(jié)合實(shí)際案例進(jìn)行�,會(huì)起到更好的培訓(xùn)效果�����。
綜上���,原始記錄是進(jìn)行了相應(yīng)研制工作的證據(jù)性文件�����,也是申報(bào)資料的依據(jù),通過記錄審核,查漏補(bǔ)缺,保證記錄和數(shù)據(jù)的真實(shí)、準(zhǔn)確��、完整��、可追溯��,確保研發(fā)項(xiàng)目通過核查、獲得批準(zhǔn)�����,所以研發(fā)人員要重視記錄審核���,規(guī)范記錄審核�。
參考資料:
GMP指南 質(zhì)量管理體系2023版
《藥品研究實(shí)驗(yàn)記錄暫行規(guī)定》2000年
《藥品記錄與數(shù)據(jù)管理要求(試行)》2020年第74號(hào)公告
《藥品注冊(cè)核查要點(diǎn)與判定原則(藥學(xué)研制和生產(chǎn)現(xiàn)場(chǎng))(試行)》2021年第30號(hào)通告