編者按
中藥已經(jīng)在中國使用了數(shù)千年��,并在維護人體健康方面發(fā)揮關(guān)鍵作用�。全球普遍開始尋求自然�����、整體的疾病治療方式�,世界范圍內(nèi)對中藥的需求穩(wěn)步增長�。隨著人們對中藥復(fù)雜性認識的加深,為了更好地認知中藥的化學(xué)成分,多成分含量測定已成為中醫(yī)藥領(lǐng)域科學(xué)家的共識�。
中國工程院院刊《Engineering》2019年第1期刊發(fā)中國科學(xué)院上海藥物研究所研究員果德安研究團隊的《對良好中藥標準的深層化學(xué)認知》一文�����。文章系統(tǒng)總結(jié)了中藥非靶向和靶向分析化學(xué)的最新技術(shù)和方法學(xué)進展,并描述了應(yīng)用一標多測法和一法多用法兩種制定中藥質(zhì)量標準的質(zhì)量控制策略�。
一、引言
中藥已經(jīng)在中國使用了數(shù)千年,并在維護中國人身體健康方面發(fā)揮了關(guān)鍵的作用。隨著全球普遍開始尋求自然����、整體的疾病治療方式�,世界范圍內(nèi)對中藥的需求穩(wěn)步增長��。這使得制定國際認可的質(zhì)量標準比人類歷史上任何時候更為重要��。藥品的主要質(zhì)量標準以藥典中的說明為主。現(xiàn)今���,《中國藥典》《美國藥典》和《歐洲藥典》越來越重視增加草本藥物相關(guān)專論的數(shù)量和提高相關(guān)專論的水平。中藥正在走向全球化發(fā)展道路�����,《歐洲藥典》(9.2版)中共收載了81種中藥材標準�����,《美國藥典》(41版)中共收載了37種植物藥及其制劑����。
更好地了解中藥的化學(xué)成分可以提高中藥整體質(zhì)量控制的水平���,現(xiàn)在應(yīng)用創(chuàng)新質(zhì)量控制技術(shù)使應(yīng)對這些挑戰(zhàn)更具有可行性�。盡管最新版的《中國藥典》(2005版)中收錄了618種中藥材和中藥飲片,但這些專論仍缺少重現(xiàn)性好的整體質(zhì)量標準���,有的甚至缺乏明確的定量指標。因此,可以看出傳統(tǒng)質(zhì)量控制模型不適用于復(fù)雜的中藥質(zhì)量評估。文章提出了一種系統(tǒng)的中藥質(zhì)量研究方法����,即在全面�����、系統(tǒng)的研究基礎(chǔ)上發(fā)展普遍適用的��、簡化的標準�����。就本方法而言,更深入地認識中藥化學(xué)性質(zhì)是理解和轉(zhuǎn)化中藥標準最重要的一環(huán)��。這是因為化學(xué)成分是中藥的治療基礎(chǔ)�,因此與中藥質(zhì)量有著不可分割的緊密聯(lián)系。更深入地了解化學(xué)成分情況也有利于揭示中藥質(zhì)量研究的四大挑戰(zhàn)和制定質(zhì)量標準:①中藥植物中化學(xué)成分的分析和表征���,尤其中藥復(fù)方中所含的化學(xué)成分;②單一成分質(zhì)量控制方法不適用于多成分體系的中藥�,迫切需要制定全面的質(zhì)量控制模型�����;③需要闡明中藥中的活性成分,甚至有效成分�;④需要制定可廣泛應(yīng)用的科學(xué)的��、實用的、可行的質(zhì)量標準�����。如丹參包含兩種主要的化學(xué)成分:正是基于在20世紀30年代首次發(fā)現(xiàn)的丹參酮和依靠20世紀80年代分離技術(shù)的發(fā)展分離出的丹酚酸?��!吨袊幍洹罚?990版)首次將丹參酮IIA列為一種鑒定標記,《中國藥典》(2005版)將丹酚酸B選作一種定量指標���。隨著更深入地了解丹參的化學(xué)性質(zhì)和質(zhì)量控制方法的進展,利用丹參酮IIA同時定量其他兩種丹參酮——隱丹參酮和丹參酮I�����,以便提高整體質(zhì)量控制��,同時保持檢測的總成本?!睹绹幍洹罚?8版)和《中國藥典》(2015版)中已經(jīng)收錄了最新的丹參專論�。最近�,提出了一種新的中藥質(zhì)量標準物(Q-marker)概念,這將改變中藥質(zhì)量控制模式的范式�����。它被定義為一種中藥的天然����、可分析、功能性可追蹤的化學(xué)成分。這種中藥質(zhì)量標準物概念也體現(xiàn)了深入了解各種中藥化學(xué)成分的重要性。
自1997年以來�����,液相色譜(LC)-質(zhì)譜(MS)聯(lián)用技術(shù)已廣泛應(yīng)用于中藥化學(xué)成分的深入分析��。一種中藥的成分包含數(shù)千種初級代謝產(chǎn)物和次級代謝產(chǎn)物�。在大多數(shù)情況下�,分析研究均著重于次級代謝產(chǎn)物——分子量在2000 Da以下的小分子。色譜分離和結(jié)構(gòu)解析等傳統(tǒng)的植物化學(xué)技術(shù)仍是獲得有關(guān)中藥化學(xué)成分復(fù)雜性準確信息的金標準。但是��,若要快速了解中藥的化學(xué)組成�,采用化學(xué)成分分離的方法就過于繁瑣耗時。將MS技術(shù)與色譜技術(shù)結(jié)合的聯(lián)用分析技術(shù)的發(fā)明提供了一種非常強大的手段�,可以詳盡分析中藥中的微量成分���,并通過單次實驗提供中藥化學(xué)復(fù)雜性的鳥瞰視圖�。就這一點而言����,超高效液相色譜(UPLC)-MS聯(lián)用技術(shù)和超臨界流體色譜(SFC)-MS聯(lián)用技術(shù)均是非常有效的手段。與簡單的色譜儀相比�����,科學(xué)家借由此類聯(lián)用儀器可以得知中藥中含有多少種成分�,哪些成分可以提供準確的質(zhì)荷比(m/z)���、有規(guī)律的離子碎片信息和離子的碰撞橫截面積(CCS)�����。過去5年來���,隨著LC-MS聯(lián)用技術(shù)的飛速發(fā)展��,智能地探索中藥中數(shù)千種化學(xué)成分和定量數(shù)百種分析物成為可能。這種分析能力開辟了深入認識中藥化學(xué)性質(zhì)的全新紀元��,并開啟了結(jié)構(gòu)解析的智能大數(shù)據(jù)時代�����。應(yīng)當(dāng)注意的是���,探索大量化學(xué)成分應(yīng)與整體質(zhì)量和(或)功能活性相關(guān)聯(lián)��,且應(yīng)通過智能數(shù)據(jù)挖掘為中藥質(zhì)量標準提供有用信息。
隨著對中藥高度復(fù)雜性的認識逐漸加深,多成分含量測定已成為中藥科學(xué)家的共識�。但是����,在中藥制藥行業(yè)實踐中應(yīng)用多成分質(zhì)量控制有兩個難題:一是對照物質(zhì)成本高昂����;二是在中成藥質(zhì)量控制中���,常常同一成分需要采用不同的處理方法,導(dǎo)致檢測工作耗時耗力���。針對第一個難題,提出了一標多測(SSDMC)方法���,該方法將廣泛應(yīng)用于《中國藥典》(2020版),為了解決第二個問題�����,提出了一法多用法���,可用于通過相同的樣品制備和分析方法定性或定量不同中成藥中的多個相同成分�����。
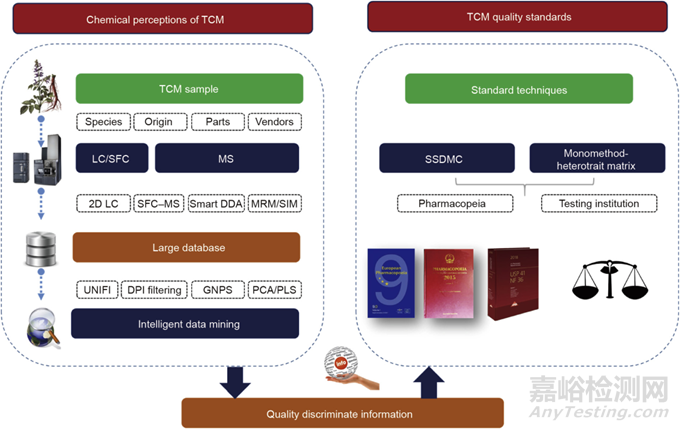
2009年��,應(yīng)用液質(zhì)聯(lián)用技術(shù)系統(tǒng)分析了中藥中不同次級代謝產(chǎn)物分析方法的應(yīng)用。Zhang等綜述了質(zhì)普分析法在中草藥的體外和體內(nèi)應(yīng)用�,He等報道了1997—2005年采用液質(zhì)聯(lián)用技術(shù)進行中藥分析的文獻計量學(xué)評價�����。該文章主要涵蓋過去5年內(nèi)深入探索中藥化學(xué)成分的新技術(shù)的發(fā)展,包括二維(2D)LC-MS聯(lián)用技術(shù)�、儀器依賴型數(shù)據(jù)采集法����、儀器非依賴型數(shù)據(jù)采集法���、通過LC-MS聯(lián)用技術(shù)進行多成分定量測定����、SFC-MS聯(lián)用技術(shù)的新應(yīng)用以及智能數(shù)據(jù)挖掘法�����。文章還提出了兩種新的質(zhì)量控制方法來建立完善的中藥質(zhì)量標準(圖1)�����。
圖1 良好中藥質(zhì)量標準的深層次中藥化學(xué)認知
二、關(guān)于深入了解中藥化學(xué)性質(zhì)和質(zhì)量標準的理念
(一)關(guān)于深入了解中藥化學(xué)性質(zhì)的理念
利用物質(zhì)信息數(shù)據(jù)庫對中藥進行全息描繪是加深中藥化學(xué)性質(zhì)理解的基本思路。如上所述��,中藥通常包含數(shù)千種化學(xué)成分��。這些成分包括有關(guān)該中藥質(zhì)量和功能活性的所有信息��。因此,首先要探討的是以下3個問題:①中藥中含多少種化學(xué)成分�?②這些化學(xué)成分的結(jié)構(gòu)是什么?③這些化學(xué)成分的含量是多少����?然而�,即使這些問題相對簡單,仍然無法通過對某些類型的代謝產(chǎn)物使用現(xiàn)代分析技術(shù)來完全解答這些問題���。
幸運的是,對于被廣泛認作主要活性物質(zhì)的主要次級代謝產(chǎn)物��,如香豆素����、皂苷、生物堿和黃酮類�,已經(jīng)報道了許多創(chuàng)新技術(shù)����,這些技術(shù)可以更好地應(yīng)對上述3個問題的挑戰(zhàn)���。多維LC-MS聯(lián)用技術(shù)是目前最重要的技術(shù)���,用于探索中藥中多樣又復(fù)雜的化學(xué)成分�,該技術(shù)包括離線和在線全二維 LC-MS分析。若結(jié)合離子淌度(IM)作為參數(shù),則可以實現(xiàn)四維分析方法�。該方法使得采用2D LC-MS聯(lián)用技術(shù)解析化學(xué)成分的理論峰容量可達9000左右�,完全滿足中藥的復(fù)雜程度�����。在沒有離子碎片信息的情況下很難表征和鑒定獲得的所有成分���。因此�����,提出了許多用于獲取二級質(zhì)譜(MS/MS)或多級質(zhì)譜(MSn)碎片信息的智能觸發(fā)數(shù)據(jù)依賴型采集(智能DDA)方法,包括儀器依賴型方法和數(shù)據(jù)后處理方法����。與LC-MS聯(lián)用法相比�����,SFC-MS用法提供了一種正交選擇性,更適合探索脂類和同分異構(gòu)體成分。針對所探索化學(xué)成分的定量信息����,已提出了廣泛靶向定量的多反應(yīng)監(jiān)測(MRM)方法���。隨著化學(xué)成分數(shù)據(jù)集的不斷積累���,應(yīng)采納智能數(shù)據(jù)挖掘方法,例如利用UNIFI軟件自動分析MS數(shù)據(jù)����、利用全球天然產(chǎn)物協(xié)會分子網(wǎng)絡(luò)(GNPS)自動對離子碎片MS/MS分類��、利用支持向量機(SVM)或神經(jīng)網(wǎng)絡(luò)(NN)技術(shù)關(guān)聯(lián)和預(yù)測質(zhì)量。
(二)關(guān)于中藥質(zhì)量標準的想法
應(yīng)認真考慮中藥質(zhì)量標準檢測的成本和效率���。對于整體質(zhì)量標準而言,對照物質(zhì)的成本是實施專論的主要障礙之一,尤其是在企業(yè)環(huán)境中�����。一標多測法提出僅通過一種對照物質(zhì)準確測定所有其他分析物的含量��,這大大降低了對對照物質(zhì)的需求��。檢測時間長是使檢測過程變得繁瑣的另一個因素,尤其是在對不同復(fù)方中藥中相同成分進行定性或定量分析時。針對定性分析的一法多用法以UPLC-QDa MS技術(shù)為基礎(chǔ),是通過采用一種低成本、緊湊的單四極桿MS技術(shù)��。在本項技術(shù)中����,通過選擇性離子監(jiān)測(SIM)方法監(jiān)測植物藥物的特征質(zhì)量成分,那么含有該成分的所有中成藥(CPM)均可使用該方法進行定性控制��。對于定量分析���,則采用2D LC多次中心切割分離(MHC)技術(shù)��。一個中藥的多個分析物可按順序轉(zhuǎn)移至第二個色譜柱�����,這將最大限度降低對紫外檢測器的干擾并通過紫外檢測器直接進行檢測。該方法也適用于含相同草本的所有中成藥�。
三�、中藥物質(zhì)基礎(chǔ)解析技術(shù)的最新進展
(一) LC-MS 聯(lián)用技術(shù)的多維分離
事實證明��,LC-MS聯(lián)用技術(shù)是一種非常實用的方法,可以揭示中藥的化學(xué)基礎(chǔ)����、新陳代謝和作用機制��。盡管在結(jié)構(gòu)、色譜柱和檢測器靈敏度方面有所改善�,但由于中藥的復(fù)雜性��,傳統(tǒng)的LC-MS聯(lián)用法仍可能存在代謝產(chǎn)物共洗脫的問題,尤其是微量分析物和高豐度分析物之間�。目前已提出了通過增加額外的分離維度(即2D LC和IM-MS)來提高性能的策略�����,尤其是在提高色譜和質(zhì)量分離方面�����。
根據(jù)兩個維度之間接口的方式,2D LC可以大致分為離線2D LC和在線2D LC�����。在離線2D LC中��,第一維分離出的各流分被收集����、處理和重新注入第二維中��。此過程自動化程度較低����,因此未引入接口���。在線2DLC分析中引入了一個特殊接口�����,自動將各流分轉(zhuǎn)移至第二維。根據(jù)是否所有1D洗脫液均經(jīng)過了2D分離�����,可將2D LC分為全2D LC分離和中心切割2D LC分離�。在中心切割2D LC中,只有當(dāng)前關(guān)注的那些成分轉(zhuǎn)移至第二維�����。
1. 離線 2D LC
離線2D LC不涉及溶劑的不相容問題����,因為第一維分離出的各流分在注入第二維前已經(jīng)加以濃縮和重新溶解。因此�,該系統(tǒng)體現(xiàn)了不同分離機制的正交聯(lián)用并提供了顯著增加的峰容量����。盡管因為人為介入濃縮�����、重新溶解和重新注入程序而存在樣品損失����、低效率���、低自動化和潛在污染等問題��,該系統(tǒng)仍然是對復(fù)雜中藥進行全面分析的最有效的方法。
通常,建立離線2D LC-MS聯(lián)用系統(tǒng)有4種組合模式,分別是正相(NP)×反相(RP)��、親水作用色譜(HILIC)×RP�、RP×RP、細胞膜色譜(CMC)×RP。在所有這些模式中����,RP色譜柱通常充當(dāng)?shù)诙S色譜柱�,因為RP色譜柱經(jīng)觀察具有高峰容量且與MS良好相容���。
(1)NP×RP:由于其獨特的保留機制��,NP色譜柱是RP色譜柱良好的替代品��。蟾蜍皮是最著名的中藥之一,具有很強的抗腫瘤活性�。Zhang等提出了一種根據(jù)第一維的XAmide色譜柱和第二維的XUnion C18 色譜柱檢測蟾蜍甾二烯內(nèi)酯的全離線2D NP/RPLC-MS聯(lián)用法���。通過幾種面積算法�,利用15種蟾蜍甾二烯內(nèi)酯混合物研究了該系統(tǒng)的正交性,研究結(jié)果正交性達到49.6%。最終����,在蟾蜍皮中鑒定處理64種蟾蜍甾二烯內(nèi)酯��,包括33種微量成分和11對異構(gòu)體。該團隊還提出采用其他兩種離線二維系統(tǒng)(HILIC×RP和RP×RP)進行蟾蜍甾二烯內(nèi)酯的制備法。
(2)HILIC×RP:盡管NP顯示與RP色譜柱存在高正交性�����,但由于其溶劑不環(huán)保且應(yīng)用范圍有限��,即使在2D LC系統(tǒng)中也未得到廣泛利用�。HILIC通常被視為“高度含水的NP”色譜�����,以乙腈和水為主要流動相。和RP不同,水是強效洗脫溶劑。2016年,Jin等綜述了HILIC固定相的最新進展和應(yīng)用���。HILIC顯示了與常規(guī)RP固定相的互補分離,適用于糖苷�����、低聚糖��、類固醇和酚酸�。這種模式的重點在于HILIC用作第一維色譜柱的順序�����。這種模式已成功應(yīng)用于人參屬的人參皂苷���、紅花的查耳酮碳苷和黃酮類O-糖苷��、丹參(根莖,Salvia miltiorrhiza)的酚酸以及銀杏(Ginkgo biloba)葉提取物的成分���。為了表征人參根莖和人參葉中的人參皂苷,提出了結(jié)合XBridge Amide色譜柱(1D)和亞乙基橋雜化(BEH)-C18 色譜柱的2D LC系統(tǒng)���,該系統(tǒng)與線性離子阱四極桿(LTQ)-Orbitrap系統(tǒng)連接。此離線2D LC系統(tǒng)的正交性為69%����,峰容量可達到8925�����。共檢測出646種人參皂苷,其中427種為潛在的新代謝產(chǎn)物��,而在2012年之前報道11種人參中僅有289種皂苷�。在2018年�����,利用相同的2D LC系統(tǒng)并結(jié)合新的數(shù)據(jù)依賴性采集(DDA)技術(shù)��,首次在三七(根莖,P.notoginseng)中篩查出945種人參皂苷�,揭示了662種潛在的新型人參皂苷(圖2)��。在這種模式下,分析物特點是具有中等極性����,可以保留在HILIC和RP中����。
圖2 利用離線2D LC-高分辨質(zhì)譜(HRMS)聯(lián)用技術(shù)和基于UNIFI軟件預(yù)測代謝產(chǎn)物數(shù)據(jù)匹配對三七(P. notoginseng)葉(PNL)進行全局分析的典型流程圖
(3)RP×RP:此模式首次應(yīng)用pH正交性�����。該模式由酸性條件流動相(1D)與帶正電荷的RP色譜柱(Acchrom XCharge C18)和堿性流動相(2D)與常規(guī)反相色譜柱(EVO C18 )組成�。利用此模式,首次系統(tǒng)解析了《中國藥典》(2015版)中收錄的鉤藤(鉤藤屬)5種植物來源的吲哚類生物堿����。離線2D LC系統(tǒng)的正交性為74%��。有效地顯示和表征了鉤藤5種植物來源的1227種吲哚類生物堿,表明該物種具有較高的化學(xué)多樣性��。中藥中生物堿成分更適用于這種模式�����。
(4)CMC×RP:CMC是一種生物親和性色譜��。具有特定受體的細胞膜吸附在活性硅表面��,形成細胞膜固定相��。只有與受體高度親和的成分才得以保留。將細胞膜色譜作為第一維構(gòu)建2D LC系統(tǒng)有利于克服色譜柱壽命短����、效率低��、峰容量低以及結(jié)構(gòu)鑒定效率低的缺陷。Yue等利用自制的β-1腎上腺素能受體(β1 AR)/CMC柱分析了黃連(Coptis chinensis)提取物,并通過UPLC-MS聯(lián)用技術(shù)分析了各流分。最后����,鑒定黃連堿是抑制β-1 AR的主要活性成分���。通過體外藥理學(xué)試驗進一步證實了該結(jié)果����。此模式可以直接篩出有效的活性成分���。
2. 在線 2D LC
在線2D LC分析中引入了一個特殊接口���,將各流分自動轉(zhuǎn)移至第二維色譜柱�。中藥全在線2D LC-MS分析的1D分離時間較長(1~2 h,為了提高分辨率)��,而2D分離時間較短�。因此,該方法的峰容量通常小于離線2D LC-MS聯(lián)用法的峰容量��;但是����,其重復(fù)性優(yōu)于離線2D LC-MS的重復(fù)性,且該方法更適用于多個樣品的分析�����。
(1)接口創(chuàng)新:在全在線2D LC系統(tǒng)中�����,接口是將所有流分從第一維自動轉(zhuǎn)移至第二維最重要的一環(huán)。HILIC×RP系統(tǒng)也在在線2D LC系統(tǒng)中顯示了良好的正交性��;但是���,在注入2D色譜柱前需要對1D洗脫液進行稀釋�,以便消除溶劑效應(yīng)。為了分析丹參中的酚酸���,選擇反沖捕捉柱作為最佳接口,包括可調(diào)節(jié)分流器���、100 μL樣品環(huán)管和100 μL溶劑混合器。從丹參中成功分離和檢測到196個色譜峰。該系統(tǒng)的正交性高達73%����。
(2)色譜柱和流動相創(chuàng)新:正交性和1D色譜柱的有機相強度是在線2D LC-MS聯(lián)用的主要考慮因素�����。Zhou等提出了一種新開發(fā)的苯基/四唑硫醚(PTAS)鍵合固定相,用于構(gòu)建RP×RP 2D LC系統(tǒng)�����。PTAS色譜柱(1D、2.1 mm × 150 mm、5 μm)與亞乙基橋雜化-C 18色譜柱(2D、3 mm × 50 mm�����、1.7 μm)的選擇性差異很大�����,主要在于其正交性可達93.2%,且疏水性較弱使其與C 18 色譜柱相容����。該系統(tǒng)用于分析莪術(shù)(根莖����,Curcuma kwangsiensis)��,該中藥共檢測出439個色譜峰(正/負離子模式)���,初步鑒定出105種化合物��,包括73種以前未報告的代謝產(chǎn)物?����;旌夏J降墓潭ㄏ噙€有利于提高正交性��。與SAX-PFP×C 18 系統(tǒng)相比���,改進的SAX-CN×C 18 系統(tǒng)在分析白花蛇舌草(全草��,Hedyotis diffusa)和半枝蓮(全草,Scutellaria barbata)時顯示出更好的峰分布情況和更合理的分析時間�����。此外����,在在線2D LC-MS系統(tǒng)中�����,可將細胞膜色譜柱用作1D色譜柱��。使用該模式成功地從附子(根,Aconitum carmichaelii)中發(fā)現(xiàn)了16種抵抗阿霉素(DOX)誘導(dǎo)心力衰竭的潛在活性生物堿成分,并在黃芩(根,Scutellaria baicalensis Georgi)經(jīng)口給藥后的大鼠含藥血清中發(fā)現(xiàn)了3種潛在的抗肝癌成分——漢黃芩素��、千層紙素A和黃芩新素��。
對于在線全2D LC�,在1D分離中采用超低流速將最大限度降低轉(zhuǎn)移量�����,并增加每個1D分離色譜峰的采樣�����,從而可在2D分離中進行多次切割。因此�,為了分析甘草的次級代謝產(chǎn)物���,在第一維中使用微徑SeQuant ZIC-HILIC色譜柱(1 mm×50 mm��、3.5 μm),以便在降低洗脫強度的情況下降低第一維中的分離速度和實現(xiàn)最小的轉(zhuǎn)移量��。最后��,檢測出89種成分���,有趣的是�,根據(jù)檢測到的特定化合物可以區(qū)分不同地理位置的甘草樣品。對于RP×RP 2D LC系統(tǒng)��,可以通過優(yōu)化流動相中的有機溶劑來提高正交性��。在分析甘草中的酚類化合物和三萜皂苷時,利用同步梯度模式可以提高色譜分離,并且在40 min內(nèi)共檢測出311種化合物����。峰容量為1329����,正交性為79.8%��。同樣地���,使用全RP×RP 2D LC-MS系統(tǒng)從皂莢(豬牙皂�,Gleditsia sinensis)中分離和鑒定出72種三萜皂苷���。
(3)結(jié)合多中心切割(MHC)技術(shù):由于中藥錯綜復(fù)雜����,在全2D LC-MS分析后通過MHC技術(shù)對1D分離出的部分洗脫液進行再次分離。MHC可通過一系列環(huán)管收集�、保留和分析連續(xù)的流分�,該環(huán)管的采用可使第二維分離時間長達3~5 min�。使用該技術(shù)可在全2D LC分析后發(fā)現(xiàn)更多成分。使用該技術(shù)分析了兩種中成藥葛根清涼湯(GQD)和燈盞生脈膠囊(DZS)。對于葛根清涼湯,使用與全2D液相色譜中相同的色譜柱結(jié)構(gòu)����,在4.4 min時將1D洗脫液裝入11個40 μl的環(huán)管���。通過2D分離連續(xù)分離這11種餾分����,共分解出13種其他的化合物�。對于燈盞生脈膠囊��,則將另一種手性高效液相色譜(HPLC)柱作為MHC的2D色譜柱�,共分離出另外12對具有良好分離度的異構(gòu)體�。該技術(shù)也可用于去除主要化合物,以探索是否存在更多微量元素��。通過這種方法��,在35 min內(nèi)分別從葛根(根�,Pueraria lobata)和粉葛(根�����,Pueraria thomsonii)的提取物中分離出271個和254個峰�����。該技術(shù)還可用于解決中成藥多成分含量測定的難題。
3. 離子淌度
離子淌度光譜儀是帶電離子在電場的作用下通過離子遷移池中惰性氣體實現(xiàn)分離��。作為確定CCS的關(guān)鍵參數(shù)����,離子的漂移時間取決于離子的大小、形狀和電荷�。CCS是給定化合物在特定條件下的物理性質(zhì)����。因此����,它是構(gòu)建庫的重要參數(shù)。色譜分離(s)��、離子遷移(μs)和質(zhì)量檢測(ms)的不同時間尺度使其可以串聯(lián)工作�����。在三者串聯(lián)時,除LC-MS分析的維度外還新增了一個維度��,該維度有利于分離中藥�、代謝體和蛋白質(zhì)組等復(fù)雜系統(tǒng)中的異構(gòu)化合物。
Tose等通過UPLC結(jié)合IM-MS聯(lián)用技術(shù)從大麻(Cannabis sativa)����、印度大麻制劑和大麻植株花葉中分離出大麻素類異構(gòu)體�。利用改良型MOBCAL計算了大麻素類異構(gòu)體的CCS,并將其與行波離子遷移質(zhì)譜(TWIM-MS)結(jié)果相關(guān)聯(lián)��。Pacini等使用超高效液相色譜(UHPLC)-紫外(UV)-TWIM MS聯(lián)用技術(shù)分析了微藻樣品中的色素��。色素在450 nm處通過特征吸收突出顯示��,接著通過TWIM-MS進一步分解和鑒定了31種不同的色素,而通過UHPLC-UV-MS聯(lián)用技術(shù)僅發(fā)現(xiàn)了26種色素����。這些表征結(jié)果也有利于區(qū)分小球藻(Chlorella vulgaris)�、杜氏鹽藻(Dunaliella salina)和三角褐指藻(Phaeodactylum tricornutum)等不同微藻物種�。Wang等在分析梔子(Gardenia jasminoides)果實時通過IM-MS實現(xiàn)了對異構(gòu)體藏紅花素-3和藏紅花素-4的分離。Willems等根據(jù)主要產(chǎn)物離子的強度區(qū)分了咖啡?����?鼘幩?��,并通過電噴霧離子化(ESI)-高場不對稱波形離子遷移譜(FAIMS)-MS技術(shù)對其進行了分離�����。采用耗時不到1 min的新方法分離和鑒定了蘋果汁/梨汁樣品中的單咖啡??鼘幩?��。Zhang等提出了一種多級MS方法���,通過整合源內(nèi)碰撞誘導(dǎo)解離(ISCID)和時間排列平行碎裂提供母離子和子離子之間更可靠的碎裂關(guān)系����,接著他們使用該方法研究了多環(huán)多異戊烯基取代間苯三酚類化合物(PPAP)的碎裂特性����。從山竹子(果實,Garcinia oblongifolia)的粗提物中共檢測出140種PPAP�����,其中7對共洗脫����、同分異構(gòu)體的PPAP是通過UHPLC-IM-MS聯(lián)用技術(shù)分化出。2D LC系統(tǒng)串聯(lián)IM-MS構(gòu)建四維分離系統(tǒng)���,并用于分析銀杏(Garcinia oblongifolia)、白花蛇舌草和半枝蓮的植物提取物。在第二維中每種化合物均只呈現(xiàn)一個峰�,對數(shù)調(diào)制時間為4 min��。在IM的幫助下,可以根據(jù)高分辨質(zhì)譜(HRMS)上的不同分子形狀區(qū)分相同的m/z離子,而在沒有IM的情況下不可能實現(xiàn)這一點。
(二) LC-HRMS 聯(lián)用技術(shù)的智能觸發(fā)數(shù)據(jù)依賴型采集(DDA)模式
利用多維分離可以發(fā)現(xiàn)更多中藥化學(xué)成分,但是���,若沒有m/z和離子碎片信息,將無法鑒定大多數(shù)化學(xué)成分。隨著HRMS技術(shù)的廣泛使用����,離子碎片采集成為最重要的一環(huán)����。通過DDA和數(shù)據(jù)非依賴型采集(DIA)可以獲取離子碎片信息�。一些文章已經(jīng)對這兩種模式進行了討論,但大部分中藥成分篩查研究傾向于根據(jù)DDA模式智能獲取離子碎片��。獲得的靶離子碎片越多�����,破譯的中藥化學(xué)成分越多。最近總結(jié)了利用LC-MS聯(lián)用技術(shù)根據(jù)特征碎片信息分析微量成分的研究,歸納了獲取更多碎片的新型DDA策略����。
根據(jù)離子強度�����、質(zhì)量包含列表或排除列表�����、同位素模式、擬中性丟失(NL)和質(zhì)量虧損觸發(fā)DDA模式。為了深入解析中藥的成分類型,這些智能觸發(fā)DDA可分為儀器依賴型和儀器非依賴型采集����。
1. 儀器依賴型智能DDA模式
大多數(shù)儀器依賴型智能DDA模式均依賴于所使用的儀器����,如LTQ-Orbitrap系統(tǒng)或離子阱(IT)-飛行時間(TOF)系統(tǒng)。最近提出了3種新方法,即NL觸發(fā)MS3(NL-MS3)法�����、質(zhì)量標簽觸發(fā)DDA�����、根據(jù)逐步母離子列表(PIL)光柵質(zhì)量虧損過濾(MDF)觸發(fā)DDA�����。
這些方法中�,NL-MS3法的使用頻率最高。該方法已用于鑒定具有相同殘基或結(jié)合物的特定類別的化合物���,如黃酮類O-糖苷、丙二?��;Y(jié)合物和脂肪酸結(jié)合物。NL-MS3法旨在利用特定的NL列表確認二羧酸結(jié)合的蟾毒素(DACB)���,通過篩查高能C-阱解離(HCD)下的各種側(cè)鏈(二羧酸)可獲得該列表。最后�,在蟾酥(Venenum bufonis)中共發(fā)現(xiàn)了78種DACB成分���,其中68種為潛在的新化合物�。
質(zhì)量標簽觸發(fā)DDA已用于研究人參提取物中的丙二?����;藚⒃碥?���。通過利用丙二?�;藚⒃碥盏乃槠?guī)律��,其中丙二酰基人參皂苷的負離子模式碰撞誘導(dǎo)解離(CID)更容易消除CO2 (43.9898 Da)�����,采用了ISCID����。然后��,啟用“質(zhì)量標簽”功能�,使用LTQ-Orbitrap質(zhì)譜儀確定MS1中的m/z值差異���。如果全掃描譜(離子強度高于閾值)中存在與定義的質(zhì)量標簽一致的質(zhì)量對�����,質(zhì)譜儀將關(guān)閉ISCID能量并觸發(fā)選中離子對的多級激活MS/MS��。這樣,只有在MS1中離子碎片質(zhì)量差值等于43.9898Da的成分才能獲得MS2�����。接著,結(jié)合NL觸發(fā)MS3法確認丙二?����;藚⒃碥?。該策略更智能且避免了復(fù)雜的數(shù)據(jù)處理過程。
逐步母離子列表(PIL)光柵MDF觸發(fā)DDA模式旨在篩查鉤藤(Uncariae Ramulus cum Uncis)5種植物來源的吲哚類生物堿[圖3(a)]�����。通過線方程和LTQ-Orbitrap中的最佳母離子質(zhì)量寬度(PMW)參數(shù)創(chuàng)建理論吲哚類生物堿的逐步PIL��。根據(jù)實驗室建立的鉤藤生物堿庫���,以整數(shù)質(zhì)量為x軸����、以質(zhì)量虧損為y軸建立方程�����。PMW經(jīng)過優(yōu)化,可以覆蓋潛在的吲哚類生物堿并盡可能排除其他成分���。與傳統(tǒng)的MDF法相比,這種方法非常方便,可以鑒定更多潛在的新吲哚類生物堿�����。
圖3 使用(a)逐步PIL光柵MDF掃描法和(b)多邊形MDF法篩查鉤藤中吲哚類生物堿的過程圖示
在這3種智能DDA方法中����,NL-MS3法適用于糖苷類或脂類成分,而質(zhì)量標簽觸發(fā)DDA法僅適用于在ISCID中存在固定質(zhì)量差異的成分。最后一種DDA模式適用于在波動范圍較小的情況下MDF和質(zhì)量之間存在線性關(guān)系的成分。
2. 儀器非依賴型智能DDA
母離子列表法(PIL)觸發(fā)DDA模式通常不依賴儀器�����,這意味著這類模式可用于大多數(shù)高分辨串聯(lián)質(zhì)譜儀��。為了智能地生成包含特定種類成分的PIL����,目前已提出了3種新方法�����,分別是多邊形MDF法����、源內(nèi)多次碰撞(IMC)-中性丟失過濾法(NLF)和同位素模式精過濾法(FIPF)����。
通常�,通過軟件實現(xiàn)MDF,如AB Sciex PeakView����、Waters MetaboLynx XS�、Thermo Scientific MetWorks和Microsoft Excel���。目前已經(jīng)相繼設(shè)計了經(jīng)典矩形MDF法和改良MDF法等不同算法�,目的是在降低假陽性和擴大靶覆蓋范圍之間找到可接受的折衷方案。MDF法已用于通過高分辨質(zhì)譜數(shù)據(jù)選擇性地檢測�����、提取或分析關(guān)注的成分�����。MDF法可有效鑒別中藥的不同化學(xué)成分���。提出了兩種不同于常規(guī)矩形繪圖算法的多邊形MDF法����。在篩查三七(P. notoginseng)中的皂苷時,設(shè)計了五邊形繪圖算法來挑選當(dāng)前關(guān)注的母離子。在篩查華鉤藤(Uncaria sinensis)中的吲哚類生物堿時,提出了多邊形MDF法[圖3(b)]���。這種算法以各鉤藤生物堿庫和分子設(shè)計為基礎(chǔ)。鉤藤中所有潛在的吲哚類生物堿均涵蓋在質(zhì)量范圍-質(zhì)量虧損范圍內(nèi)(質(zhì)量范圍指觀察值m/z的整數(shù)部分),并表現(xiàn)為由8個頂點描繪的八角形區(qū)域�����;然后可以根據(jù)全MS總離子列表的限制生成PIL����,以確定吲哚類生物堿的種類��。
IMC-NLF法用于生成人參中丙二?���;藚⒃碥盏腜IL�����。該方法利用丙二?�;藚⒃碥盏乃槠?guī)律,即以低能量碎裂產(chǎn)生CO2或丙二?;?��。首先���,使用多源碰撞能量獲取全掃描原始數(shù)據(jù)��,處理數(shù)據(jù)并將其導(dǎo)出至逗號分隔值(CSV)表格中。然后��,使用NL-MS檢測器軟件生成PIL��。最終從人參中提取和鑒定了69種丙二?�;藚⒃碥铡?/span>
使用FIPF法篩查中藥中硫衍生物�,在m/z=400時將分辨率設(shè)定為100 000半峰全寬(FWHM)����。硫磺熏蒸中藥是一個關(guān)鍵問題�����,因為這種中藥能產(chǎn)生具有未知生物效應(yīng)的硫衍生物。但是��,很難通過傳統(tǒng)的同位素模式過濾法(IPF)找出新的硫衍生物�����,因為天然穩(wěn)定的 34S不如 37Cl和 81Br豐度高���,也就是說��, 34S信號將受 13C2 + 18O貢獻的巨大影響。根據(jù)M+2同位素組成(12Cx1Hy16Oz32S 13C218O���、12Cx+21Hy16Oz+134S),硫衍生物的M+2差異為Δm=0.0098 Da�。采用這種細化的標準����,從葛根和粉葛中初步鑒定出9種硫衍生物����,并且快速比較了55種市售樣品,這表明可以從更多葛根樣品中鑒定出硫磺熏蒸過程�����。
在上述3種方法中����,多邊形MDF法適用于大多數(shù)成分為相同類型的樣品����。IMC-NLF法可用于通過DIA法獲取的大量原始數(shù)據(jù)�。而FIPF法僅適用于具有特殊元素的成分�����。大多數(shù)PIL觸發(fā)DDA法會與動態(tài)排除(DE)結(jié)合使用�����,如葛根、陳皮葉(Citrus reticulata)和香砂六君子加減顆粒分析所示�����。應(yīng)該指出的是���,MS實驗過程中質(zhì)量列表過長可能導(dǎo)致工作緩慢和數(shù)據(jù)丟失�����,PIL可以拆分為兩個或兩個以上的分列表�����。Shen等利用兩個各含約250種化合物的分列表表征了丹參中的190種聚合酚酸。
(三)使用 LC-MS 聯(lián)用技術(shù)進行靶分析
使用LC-MS聯(lián)用技術(shù)進行多維分離通常用于化學(xué)成分的非靶向分析。但是,靶分析同樣重要,因為靶分析可以提供更可靠的中藥定量信息或更具體的質(zhì)量特征相關(guān)信息。目前存在兩種主要的采集模式:MRM和SIM�����。上述兩種方法已得以廣泛應(yīng)用����,但是,鑒于中藥的復(fù)雜性,本章也提及了一些創(chuàng)新方法���。
1. 多反應(yīng)監(jiān)測
MRM法應(yīng)用三重四極桿LC-MS/MS聯(lián)用系統(tǒng)��,同時監(jiān)測每種分析物的特定母離子和特征產(chǎn)物離子��,現(xiàn)已成為廣泛用于同時對多種低豐度代謝產(chǎn)物進行定量研究的常用方法。然而,MRM法存在一些瓶頸,如濃度范圍��、極性范圍��、一次實驗中離子對數(shù)量有限以及缺乏真實的對照物質(zhì)��。為了解決甘草屬物種(根莖)中成分的濃度范圍問題,采用7種方法對3個來源的樣品中151種生物活性次級代謝產(chǎn)物進行了定量分析����,通過UPLC/MRM法確定了微量成分����。通過DNA條碼的相關(guān)性分析����,發(fā)現(xiàn)甘草雜交可能顯著改變甘草的化學(xué)成分,而且父本對后代的貢獻大于母本�����。若極性范圍廣或存在對映異構(gòu)體����,則在連續(xù)通過模式或中心切割2D模式下采用兩個不同的色譜柱。在連續(xù)通過模式下使用RP LC和HILIC的色譜柱最終鑒定了肉蓯蓉(全草��,Cistanche salsa)中的21種化合物��,并在中心切割2D模式下結(jié)合非手性-手性柱同時對18種香豆素進行定量分析��,包括前胡(根,Peucedanum praeruptorum)的7對對映異構(gòu)體。定時MRM法或動態(tài)MRM法可用于增加同時檢測的離子對數(shù)量,從而可在提高靈敏度�、優(yōu)化檢出限(LOD)和定量限(LOQ)的情況下鑒定更多化合物,包括牛黃上清丸中的41種成分以及人參和西洋參中的221種人參皂苷�����。而針對缺乏真實對照物質(zhì)的問題,需要新的方法來優(yōu)化MRM參數(shù)和實現(xiàn)絕對定量����。定量核磁共振氫譜(q1H-NMR)結(jié)合色譜分離用于產(chǎn)生假混合標準溶液��,從而可以對更多化合物進行定量分析。在四極桿飛行時間(QTOF)儀器上使用步進式MSAll (sMSAll)技術(shù)可以預(yù)測沒有標準值的MRM參數(shù)���,并將這些參數(shù)傳輸至三重四極桿MS系統(tǒng)。同樣地�,IT/MS的 MS2可用于生成MRM模式的主要參數(shù)�����,這些參數(shù)在定量測定中可提供快速直接的轉(zhuǎn)換設(shè)計。
2. 選擇性離子監(jiān)測
由于需要對中藥中特征化合物進行表征�����,從而為中藥質(zhì)量控制 ����、定性分析和鑒定研究提供支持數(shù)據(jù),選擇性離子監(jiān)測法應(yīng)運而生��。與MRM法相比��,SIM法在應(yīng)用ACQUITY QDa MS系統(tǒng)等緊湊型單四極桿MS時易提供合理的選擇性��、更好的適用范圍。因為可在ES– 和ES+ 之間快速切換�����、參數(shù)設(shè)置少��、成本低、占用空間少�,QDa MS是進行SIM的理想工具���。
(四)超臨界流體色譜(SFC)
SFC是一種傳統(tǒng)技術(shù)���,該技術(shù)中將具有低黏度和高擴散性的超臨界流體作為流動相����,如超臨界二氧化碳(sCO2)�。隨著技術(shù)的進步,現(xiàn)已具有更優(yōu)良的重復(fù)性和穩(wěn)定性��,使該技術(shù)更適用于中藥研究�����,如超高效超臨界流體色譜法(UHPSFC)�。該技術(shù)具有分離效率高�����、流動相速度快��、分析時間短和環(huán)境友好等特點。近年來��,已有研究對SFC在中藥研究中的應(yīng)用進行了綜述��,文章將提到部分特殊應(yīng)用�����。
1. 中藥脂質(zhì)組學(xué)
脂質(zhì)組學(xué)是對基質(zhì)中的大量脂質(zhì)進行全局分析的學(xué)科,在很大程度上依賴于分離科學(xué)技術(shù)的新發(fā)展����。SFC-MS聯(lián)用技術(shù)可以在降低成本和減少分析時間的同時提高脂質(zhì)覆蓋率,因此可以作為其他分析技術(shù)的替代方案。通過UHPSFC/QTOF-MS聯(lián)用技術(shù)綜合分析和比較了使用甲基叔丁基醚從三種人參同屬種中提取的脂質(zhì)組��。將甲醇作為改性劑�����、甲醇/0.2 mmol·L−1乙酸銨作為補充流體�����,利用1.7 μm填料粒徑的Torus 2-吡啶甲基胺色譜柱分析樣品��。成功分離了6種脂質(zhì)亞類,且與RP系統(tǒng)相比,實現(xiàn)了對極性脂質(zhì)和脂質(zhì)異構(gòu)體更好的分離�。在分析了60批人參樣品后�,共發(fā)現(xiàn)了24種三?���;视汀T摷夹g(shù)還用于探索不同產(chǎn)地薏仁的脂類標記物,在這種情況下���,二酰甘油酯可作為主要的質(zhì)量標記物。
2. 中藥的極性化學(xué)成分
除脂類之外����,還通過有效應(yīng)用SFC技術(shù)研究中藥的極性化學(xué)成分����。呋甾皂苷類化合物C-22位羥基具有活性���,且易與低級醇發(fā)生反應(yīng)��。在40 ℃時,使用甲醇(含0.2% NH3 ·H2O和3%水��,作為改性劑)在二醇基色譜柱上分離了22 min���,成功分離出10種相似的呋甾皂苷類結(jié)構(gòu)����。SFC技術(shù)也可用于分離具有相同糖苷配基和不同糖鏈的螺甾皂苷類化合物,該技術(shù)對糖苷配基中羥基的數(shù)量和位置非常敏感�����。但是�,使用UHPSFC技術(shù)分離具有不同糖苷配基和相同糖配基的螺甾皂苷類化合物并不是一種理想的手段,可以用UHPLC法取代�。UHPLC法可有效鑒別糖苷配基的變化�����,且顯示是否受糖苷配基中雙鍵的影響。將甲醇/乙腈(體積比70:30)���、1%三氟乙酸(TFA)作為改性劑,流速為0.8 mL·min−1���,使用亞乙基橋雜化色譜在25 min內(nèi)對蒼耳子(果實,Xanthium sibiricum Patr)中的8種酚酸進行了定量分析���。針對類黃酮,將0.1%磷酸甲醇溶液作為極性流動相���,使用Zorbax Rx-SIL色譜柱在15 min內(nèi)對菊花(花,Chrysanthemum morifolium Ramat)中的5種類黃酮進行了定量分析��。對于藥用真菌牛樟芝(Antrodia camphorata)中的7對25R/S-麥角甾烷 類化合物�����,Chiralcel OJ-H色譜柱(4.6 mm×250 mm、5 μm���、手性)和Princeton乙基吡啶色譜柱(2-EP、4.6 mm×250 mm��、3 μm����、非手性)具有不同的分析優(yōu)勢。手性色譜柱有效分離每對化合物���,而非手性色譜柱能有效分離不同的對,即使每個25R/S差向異構(gòu)對的分離效果不如OJ-H色譜柱的分離效果好。此外,SFC-MS聯(lián)用技術(shù)優(yōu)化了3種三萜皂苷(苦丁冬青苷、甾體皂苷和人參皂苷)。為了綜合判斷SFC技術(shù)的代謝產(chǎn)物分析性能�����,采用120種高度多樣化的天然化合物對15種不同固定相色譜柱進行了比較。研究發(fā)現(xiàn)����,3種固定相(二醇�、未封端C18 和2-EP)適用于非靶向監(jiān)測分析和方法開發(fā)��,因為這些固定相可以對120種天然化合物中的101種進行適當(dāng)洗脫��。研究發(fā)現(xiàn),未封端T3 C18 和極性P-PFP可以為特定天然分子亞類提供更大的選擇性��。
3. 制備對照化合物
制備高純度對照物質(zhì)是中藥質(zhì)量控制的關(guān)鍵����。然而,由于極性溶劑或水溶劑可能出現(xiàn)快速結(jié)構(gòu)變化���,或由于熱不穩(wěn)定性,一些天然化合物純度不高��,無法用作標準物質(zhì)�。使用無水流動相在兩個非手性Torus 1-氨基蒽(1-AA)色譜柱和Torus DIOL色譜柱上分別分離了兩對7-螺羥吲哚類生物堿����。離線2D SFC-RP LC聯(lián)用系統(tǒng)具有良好的正交性,已成功從牛蒡子(果實,Arctium lappa L.)中分離出12種木酚素��。
(五)大數(shù)據(jù)庫集的數(shù)據(jù)挖掘
通過LC-HRMS法或SFC-HRMS聯(lián)用法對中藥進行全面的非靶向分析�����,獲取高維度大型數(shù)據(jù)集��。即使與代謝組學(xué)相比,從這些大數(shù)據(jù)庫集中收集數(shù)據(jù)挖掘信息也非常困難,因為中藥的化學(xué)組成多樣性明顯多于生物內(nèi)源性化學(xué)組成。此外,很少有科學(xué)家涉足該領(lǐng)域,能夠獲取的專業(yè)數(shù)據(jù)庫屈指可數(shù)����。因此����,借助各種生物信息學(xué)策略���,試圖從大量數(shù)據(jù)集中挖掘數(shù)據(jù)�。將介紹數(shù)據(jù)挖掘的3個方面。
數(shù)據(jù)匹配:通常是第一步���,利用精確的質(zhì)量或分子式檢索化學(xué)庫,從而識別潛在的化合物���。但是,庫中儲存的化學(xué)結(jié)構(gòu)數(shù)量有限,因此利用化學(xué)庫鑒定植物代謝產(chǎn)物非常具有挑戰(zhàn)性��。代謝產(chǎn)物預(yù)測算法可以在枚舉中使用預(yù)先定義的子結(jié)構(gòu)或“構(gòu)筑塊”��,以便預(yù)測代謝產(chǎn)物中是否存在結(jié)構(gòu)成分����;這將增加可用化學(xué)庫的化學(xué)覆蓋空間���,實現(xiàn)更高效��、結(jié)構(gòu)去重復(fù)且預(yù)測更準確。該方法尤其適用于結(jié)構(gòu)多樣的糖苷分子�,如皂苷類和糖化黃酮類化合物中的糖苷分子�����,這些分子通常由與各種糖基和酰基軛合的糖苷配基組成��。借助商業(yè)軟件UNIFI或內(nèi)部軟件PlantMATM���,在庫中生成使用葡萄糖���、木糖��、鼠李糖和丙二?���;鶈卧徊叫揎椇蛢刹叫揎椀娜藚⒃碥铡@眠@種方法匹配了三七葉中的更多人參皂苷,這種方法也適用于木酚素和多酚。
數(shù)據(jù)篩選/分類:基于LC-HRMS聯(lián)用技術(shù)原始數(shù)據(jù)(包括MS1和MS/MS或MS n 數(shù)據(jù))中包含的大量信息進行數(shù)據(jù)篩選/分類仍是一項艱巨的任務(wù)���。目前,已提出許多后采集數(shù)據(jù)處理策略,用于數(shù)據(jù)篩選或分類�����。對于MS1數(shù)據(jù)�,無處不在的基質(zhì)干擾使微量成分的表征難上加難。因此,通過結(jié)合各種篩選方法��,提出了整合的篩選策略和R腳本����。該策略成功快速地從艾葉(葉����,Artemisiae argyi)的UPLC-HRMS數(shù)據(jù)集中篩查出16種甲氧基化黃酮類化合物和55種綠原酸類似物。
對于MS/MS數(shù)據(jù)�,從碎片離子信息中獲取大量結(jié)構(gòu)信息�。因此�����,提出了關(guān)鍵離子過濾策略����,用于篩選和劃分黃芩中的不同黃酮類化合物�。最后,從黃芩中鑒定出132種化合物����,其中59種首次報道���。同樣地�,根據(jù)診斷產(chǎn)物離子劃分了威靈仙(根莖����,Clematis chinensis Osbeck) 中的三萜皂苷、 紫草( 根�,Arnebia euchroma)中的紫草寧和紫草呋喃以及杜仲(皮���,Eucommia ulmoides Oliv)和蒺藜(果實���,Tribulus terrestris)中的成分���。全球天然產(chǎn)物協(xié)會分子網(wǎng)絡(luò)(http://gnps.ucsd.edu)是另一種高效劃分大量MS/MS數(shù)據(jù)集的方法����,這是一個面向全社區(qū)組織的開放知識庫���,可以共享原始的�����、處理后的或鑒定的串聯(lián)(MS/MS)MS數(shù)據(jù)。該平臺可用于根據(jù)改良的余弦評分方案創(chuàng)建分子網(wǎng)絡(luò)����,這種方案可判定兩個MS/MS的相似性并使大量MS/MS數(shù)據(jù)的歸類過程可視化���;它將成為一種寶貴的中藥化學(xué)研究手段��。
對于MSn 數(shù)據(jù),可使用Mass Frontier軟件中的質(zhì)譜樹狀圖相似度過濾技術(shù)(MTSF)來鑒定化合物和獲取子結(jié)構(gòu)信息���。該技術(shù)可以篩選有用的HRMS和MSn 數(shù)據(jù),并通過計算相似度匹配分數(shù)建立未知化合物和模板化合物之間的聯(lián)系����。在分析二仙湯時����,使用該技術(shù)成功排除了不相關(guān)的離子。
對于混合模式���,不同裂解模式可以提供互補的碎片離子信息。通過在LTQ-Orbitrap中結(jié)合傳統(tǒng)的CID-MS3和HCD-MS2��,使用診斷產(chǎn)物離子(DPI)過濾法和NLF法對紅花中黃酮類O-糖苷的糖苷配基和糖分類�。
數(shù)據(jù)挖掘:利用化學(xué)計量學(xué)進行數(shù)據(jù)挖掘已成為一種廣泛使用的中藥質(zhì)量研究手段。主成分分析(PCA)和(正交性)偏最小二乘判別分析[(O)PLS-DA]是最常用的監(jiān)督和非監(jiān)督數(shù)據(jù)挖掘方法�����??梢宰R別對應(yīng)不同物種��、植物不同部分�����、不同產(chǎn)地、不同“炮制”����、不同供應(yīng)商]以及不同批次的質(zhì)量標記物���。然后���,利用人工神經(jīng)網(wǎng)絡(luò)(ANN)或支持向量機(SVM)�����,根據(jù)這些質(zhì)量標記物預(yù)測中藥的質(zhì)量。此外,可以通過主成分分析鑒定新型化合物��。在三重四極桿質(zhì)譜儀上通過12種不同NL/母離子(PRE)掃描得到了12份姜黃(根莖�,Curcuma longa)離子色譜圖。接著,PCA能夠?qū)⒕哂邢嗨芅L/PRE模式的數(shù)據(jù)進行聚集,而無需了解其分子量�����,而那些具有不同NL/PRE模式的化合物很容易被識別為可能具有新結(jié)構(gòu)的異常值�。
四、中藥質(zhì)量控制的應(yīng)用
隨著對中藥化學(xué)性質(zhì)認識的加深,可以確認哪些質(zhì)量標記物可用作中藥質(zhì)量標準���。為了節(jié)省標準物質(zhì)的使用和檢測時間,提出了兩種質(zhì)量策略���。
(一) 一標多測(SSDMC)法
SSDMC法僅需要一種標準物質(zhì)就能同時鑒定十余種化合物�。盡管該方法非常方便,但是其校正因子對UV檢測器和峰測量參數(shù)極度敏感。此外����,用于計算校正因子的對照溶液的濃度應(yīng)在合理范圍內(nèi)�。目前���,SSDMC法已廣泛用于中藥的質(zhì)量控制���,包括薏苡仁(種子�����,Coix lacryma-jobi L.)中的7種三酰甘油���、附子中的6種生物堿���、人參中的9種人參皂苷�、黃芩中的4種黃酮類化合物�、靈芝(Ganoderma)中的17種三萜類化合物、天麻塊莖(根莖��,Gastrodia elata)中的11種成分�����、丹參酮提取物中的4種丹參酮���、金銀花中的6種化合物(花�����,Lonicera)����、虎杖(根莖和根,Polygonum cuspidatum)中的4種化合物����、功勞木(枝��,Mahoniae Caulis)中的4種生物堿、紅景天(根莖和根��,Rhodiola crenulata H. Ohba)中的3種酚類化合物以及甘遂(根�,Euphorbia kansui)中的13中成分。波長程序�、蒸發(fā)光散射檢測器(ELSD)和MS檢測器已用于一標多測法中��。但是,對于質(zhì)譜檢測器�,在將該方法用作質(zhì)量標準方法前需要考慮更多影響因素����。除了SSDMC法��,通過對照物質(zhì)提取物進行定量分析(QASRE)和定量核磁共振(qNMR)光譜技術(shù)在節(jié)省標準物質(zhì)方面均具有獨特的優(yōu)勢�。每種方法各有利弊����,應(yīng)具體情況具體分析。
(二) 一法多用法
由幾種到幾十種中藥制成的中成藥是中藥的主要利用形式����。一方面,中成藥極大地豐富了中藥的應(yīng)用范圍��,促進個性化的藥物治療方案�;另一方面,中成藥形成了極其復(fù)雜的基質(zhì)��,妨礙質(zhì)量控制�����?�?紤]到中成藥的龐大數(shù)量及其質(zhì)量控制的難度,針對每種中成藥構(gòu)建定性和定量方法將非常耗時且相當(dāng)困難。因此,文章提出了一法多用法���,便于對不同中成藥中的相同中藥成分進行鑒定和定量測定。
1. 鑒定
中藥質(zhì)量控制的主要化學(xué)鑒定方法是薄層色譜法(TLC)和高效薄層色譜法(HPTLC)。TLC法和HPTLC法的靈敏度低��,峰容量差���,表明需要針對不同中成藥中的相同中藥成分開發(fā)新的方法��。因此����,這將導(dǎo)致樣品制備程序復(fù)雜�,實驗時間長,甚至造成無法實施任何鑒定方法的情況��。
紅花常用于多種中成藥中���,但因為中成藥中紅花含量較少����,大多數(shù)使用紅花的中成藥無法鑒定紅花的成分�。因此,通過UHPLC/QTOF-MS聯(lián)用技術(shù)對20批紅花進行了化學(xué)分析,并進行了熱穩(wěn)定性試驗后���,選擇6種查耳酮C-糖苷作為化學(xué)分類學(xué)標記物�����。接著,在這6個標記物的基礎(chǔ)上建立了一種靈敏且具有高度特異性的SIM方法,鑒定了28種不同中成藥的紅花成分��。令人驚訝的是����,在其中10批樣品中,這6個標記物無法全部檢出,而在其中兩批樣品中,幾乎檢測不出這6個標記物中的任意一個標記物���。同樣地,在鑒定舒胸片的3種中藥成分即三七、川芎(根莖��,Rhizoma Chuanxiong)和紅花(花)時�,《中國藥典》(2015版)中采用了3種繁瑣的TLC法。通過QDa MS檢測儀,選取覆蓋3種中藥的11個標記物來評估12批疏血通片。在一次實驗中鑒定出所有3種中藥,表明QDa MS技術(shù)擁有與QTOF-MS聯(lián)用技術(shù)相同的鑒定能力��。
2. 定量測定
不同中成藥中相同中藥的定量測定涉及復(fù)雜的樣品制備程序和案例依賴型色譜參數(shù)����。因此,需要建立一套通用的方法����,可以同時對涉及相同植物的不同中成藥進行質(zhì)量評估��。
三七是許多中成藥中常用的一種中藥。三七皂苷R1及人參皂苷Rg1、Re�����、Rb1和Rd是主要活性成分���,因此被視為標記物成分��。《中國藥典》(2015版)收錄了80多種涉及三七的中成藥�����,其中11種中成藥將三七作為君藥��。研究發(fā)現(xiàn)�����,某些中成藥沒有質(zhì)量控制或其質(zhì)量控制的少量幾個成分分析條件受色譜條件影響較大。構(gòu)建了中心切割2D LC系統(tǒng)�,同時測定8種不同中成藥中的5種標記物皂苷�。2D LC技術(shù)具有高峰容量��,因此簡化和統(tǒng)一了樣品制備方法����。從專屬性����、線性、重復(fù)性�����、穩(wěn)定性和準確性等方面對該方法進行了驗證���,結(jié)果證明該方法具有高特異性����、良好分辨率和高分析效率等優(yōu)點�����。對于那些具有相同定量指標的中成藥�����,這種統(tǒng)一的方法極大地促進了質(zhì)量控制(圖4)。
圖4 一法多用法與中心切割2D HPLC-UV聯(lián)用系統(tǒng)的圖示
五、展望
文章系統(tǒng)地總結(jié)了中藥非靶向和靶向分析化學(xué)的最新技術(shù)和方法學(xué)進展��,并描述了兩種制定中藥質(zhì)量標準的質(zhì)量控制策略。隨著LC-HRMS聯(lián)用技術(shù)���、液相色譜-固相萃取(SPE)-NMR聯(lián)用技術(shù)和SFC-HRMS聯(lián)用技術(shù)等聯(lián)用分析方法的不斷發(fā)展�,可以輕松快速地識別中藥中的更多微量成分���。針對非靶向分析��,離線 2D LC-HRMS聯(lián)用技術(shù)是最有效的方法,可用于鑒定中藥中的多種成分�,尤其是該技術(shù)與離子淌度聯(lián)用時�。在自動化和標準操作以及計算機輔助數(shù)據(jù)挖掘等方面的更多進展將提高重復(fù)性并減少該方法的數(shù)據(jù)分析時間��。應(yīng)該指出的是����,大多數(shù)非靶向研究僅關(guān)注中藥中的幾種次級代謝產(chǎn)物���,如皂苷���、黃酮類���、生物堿�����、酚酸和萜烯類,普遍認為這些產(chǎn)物與生物活性相關(guān)。但是,應(yīng)該更多地重視中藥中其他含量豐富的成分,如單寧��、多糖����、脂類和蛋白質(zhì),這些成分也可能影響藥理活性,從而影響質(zhì)量���。對于在線全2D LC-HRMS聯(lián)用技術(shù),改進第二維色譜柱的分析時間將顯著提高峰容量���。而針對中心切割2D LC方法,更好地集成接口可能使該方法更適用于中藥的質(zhì)量控制����。在LC-MS分析中����,IM技術(shù)為成分分析提供了一個額外的參數(shù)�,非常有利于鑒定。提高IM分離將使其更適用于中藥的化學(xué)分析��。在LC-HRMS分析中����,采用DDA模式最大程度地暴露中藥化學(xué)成分的二級碎片需要及其精巧的設(shè)計。然而,只有特定類型的化合物能產(chǎn)生DDA模式所需的碎片離子信息。DIA是一種生成MS/MS碎片的非靶向模式,它需要通過計算方法來解決碎片離子信息��。DIA領(lǐng)域仍需要更多創(chuàng)新方法���。實用性和覆蓋率是靶分析的兩大主要方面���。MRM的覆蓋范圍越廣���,驗證效果越好�,就越能提供更可靠的中藥質(zhì)量信息的相關(guān)信息��。SIM方法結(jié)合使用緊湊穩(wěn)健的質(zhì)譜儀�����,其實用性可能引起行業(yè)內(nèi)中藥質(zhì)量標準模式的變革。隨著SFC-MS聯(lián)用技術(shù)重復(fù)性和穩(wěn)健性的提高,一旦設(shè)計出更多專用色譜柱��,就可以將該方法用于更多化學(xué)分析中�����。R�、Python或MATLAB等計算機語言非常有利于理解和處理從LC-HRMS中獲取的大數(shù)據(jù)集。GNPS是一個非常有用的MS/MS數(shù)據(jù)分類和數(shù)據(jù)匹配平臺。在不遠的將來,人工智能(AI)必將成為數(shù)據(jù)挖掘最重要的手段����。隨著對中藥化學(xué)性質(zhì)認識的加深�,所有質(zhì)量相關(guān)化學(xué)信息均可以以全息方式儲存在物質(zhì)數(shù)據(jù)庫中����。
檢測成本和檢測時間是評估中藥質(zhì)量標準實用性的兩個重要因素。采用SSDMC法和一法多用法執(zhí)行更多應(yīng)用和標準規(guī)范。“深入研究����,淺出標準”的理念將提高中藥質(zhì)量標準�,并更容易將中藥轉(zhuǎn)化為藥品�。
改編原文:
Jinjun Hou, Jianqing Zhang, Changliang Yao, Rudolf Bauer, Ikhlas A. Khan, Wanying Wu, De'an Guo. Deeper Chemical Perceptions for Better Traditional Chinese Medicine Standards[J]. Engineering,2019,5(1): 83?97.