[摘要]自 2015 年��,藥品審評審批改革實施的系列措施為我國創(chuàng)新藥的發(fā)展構(gòu)建了良好的生態(tài)環(huán)境,創(chuàng)新藥的研發(fā)成果顯著����。近幾年�����,按照《藥品注冊核查工作程序》�、《核查要點和判定原則》等注冊核查規(guī)程和標準對創(chuàng)新藥研制和生產(chǎn)現(xiàn)場開展注冊核查過程中,發(fā)現(xiàn)了存在藥品注冊申請人與受托單位之間的職責不清�����、質(zhì)量管理體系不完善����、藥品生產(chǎn)質(zhì)量管理規(guī)范(GMP)不規(guī)范情況突出等問題。本文從化學藥創(chuàng)新藥注冊核查任務的特點進行研究�,梳理了化學藥創(chuàng)新藥注冊核查任務的基本情況��,通過對 2019—2021 年化學藥創(chuàng)新藥注冊核查中發(fā)現(xiàn)的主要問題進行歸納、總結(jié)和分析���,提出相應建議,以期為行業(yè)創(chuàng)新藥研發(fā)及注冊提供參考�����,為藥品監(jiān)管注冊核查管理工作提供借鑒��。
化學藥注冊按照化學藥創(chuàng)新藥��、化學藥改良型新藥、仿制藥等進行分類[1]�?��;瘜W藥創(chuàng)新藥系指含有新的結(jié)構(gòu)明確的�����、具有藥理作用的化合物,且具有臨床價值的藥品[2]��。2015 年��,《國務院關于改革藥品醫(yī)療器械審評審批制度的意見》 提出后,國家藥品監(jiān)督管理局(NMPA) 持續(xù)推進審評審批制度改革�����,多措并舉鼓勵創(chuàng)新[3]����。2019 年,全國人民代表大會常務委員會審議通過新修訂的《藥品管理法》,從法律層面固化藥品審評審批制度改革成果��。2020年新版《藥品注冊管理辦法》發(fā)布實施�����,設立了突破性治療藥物���、附條件批準�、優(yōu)先審評審批�����、特別審批4 個新藥上市注冊加快通道����,持續(xù)鼓勵藥品創(chuàng)新發(fā)展[4 -5]����。為了促進新藥研發(fā)、提高藥品質(zhì)量、保證公眾用藥安全����,《藥品注冊管理辦法》規(guī)定了由 NMPA食品藥品審核查驗中心(Center for Food and DrugInspection of NMPA��,CFDI)進行藥品注冊核查,旨在核實藥品注冊申報資料的真實性、一致性以及藥品上市商業(yè)化生產(chǎn)條件�����,檢查藥品研制的合規(guī)性��、數(shù)據(jù)可靠性等���。本文通過對 CFDI 承擔的 2019—2021 年度開展的 114 個(按受理號計) 化學藥創(chuàng)新藥注冊核查工作的實施情況及發(fā)現(xiàn)存在的主要問題進行收集��、整理及分析,提出相應建議,以期為行業(yè)新藥研發(fā)及注冊提供參考�����,為藥品注冊核查工作提供借鑒�。
一、化學藥創(chuàng)新藥注冊核查任務的基本情況
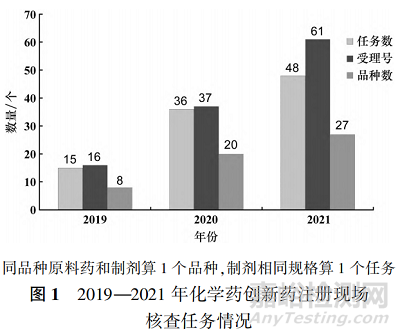
2019—2021 年,CFDI 共對 NMPA 藥品審評中心(Center for Drug Evaluation of NMPA�����,CDE) 發(fā)起的114 個(按受理號計�����,品種數(shù)為 55 個)化學藥創(chuàng)新藥注 冊 核 查 任 務 進 行 了 藥 學 研 制 和 生 產(chǎn) 現(xiàn) 場 核查[6 - 8]���。其中 2019 年任務數(shù)為 15 個(按受理號計為 16 個�,品種數(shù)為 8 個)�;2020 年任務數(shù)為 36 個(按受理號計為37 個,品種數(shù)為20);2021 年任務數(shù)為 48 個 (按受理號計為 61 個����,品種數(shù)為 27)��。圖 1顯示�,近 3 年化學藥創(chuàng)新藥的任務數(shù)量逐年遞增。因新舊法規(guī)過渡�,上述任務包括按照 2007 年《藥品注冊管理辦法》 (局令 28 號)受理的注冊申請核查任務 9 個����,2017 年 12 月《關于調(diào)整藥品注冊受理工作的公告》(2017 年第 134 號)發(fā)布后集中受理的注冊申請核查任務 63 個�,2020 年《藥品注冊管理辦法》(國家市場監(jiān)督管理總局令第 27 號)發(fā)布實施后受理的注冊申請核查任務42 個。圖2 顯示���,在核查任務省份分布上,呈現(xiàn)出明顯的區(qū)域特性��,主要集中在制藥大省�,僅江蘇、浙江�����、上海三省市的化學藥創(chuàng)新藥的任務占比就達到了 65. 7% �。在圖 3 所示的劑型分布上,以普通的口服固體制劑及注射劑為主��,片劑占比為 37% ���,注射劑占比為 11% �����,膠囊劑占比為 12% ����,此外�,原料藥占比最高,達到了 39% �����。


二����、化學藥創(chuàng)新藥注冊核查任務的特點
2.1 研發(fā)周期長,涉及研制機構(gòu)多
創(chuàng)新藥的研發(fā)周期長是業(yè)界共識���,因其“全球新”,不像仿制藥那樣有參比制劑可以進行對比研究��,一個創(chuàng)新藥的誕生需要經(jīng)歷幾年甚至十幾年的研發(fā)投入���。創(chuàng)新藥大多通過委托研發(fā)或聯(lián)合研發(fā)的方式進行����,如化學原料藥的結(jié)構(gòu)確證研究、微生物限度檢查等的方法學研究��、藥包材與藥品相容性試驗研究等��,一般需要委托具有相關能力���、資質(zhì)的機構(gòu)進行����,故一個藥品的完整研發(fā)過程常由多家機構(gòu)共同參與[9]�����。通過對 2019—2021 年共 114 個(按受理號計)化學藥創(chuàng)新藥的注冊核查任務情況進行梳理分析�,化學藥創(chuàng)新藥通常為制劑與其原料藥關聯(lián)審評�,在 CDE 啟動核查時往往藥學研制現(xiàn)場和生產(chǎn)現(xiàn)場核查同時發(fā)起,因研發(fā)周期長�����、涉及研制場地多�,在不同研制場地之間的技術轉(zhuǎn)移�、前后數(shù)據(jù)的橋接等都增加了研發(fā)的復雜性,對注冊現(xiàn)場核查來說,要核實申報資料的真實性��、一致性�����,研究周期長�����、研制場地多無疑會增加核查的難度和強度。
2.2 委托生產(chǎn)情況復雜
化學藥創(chuàng)新藥的上市申請通常涉及原料藥的關聯(lián)審評,在注冊核查中需要對原料藥的研制及生產(chǎn)現(xiàn)場進行核查。近幾年�����,我國原料藥產(chǎn)業(yè)發(fā)展之路較為曲折�����,2015 年 1 月 1 日起施行的《中華人民共和國環(huán)境保護法》����,2015 年 4 月國務院發(fā)布的《水污染防治行動計劃》��,2017 年 2 月原環(huán)境保護部等發(fā)布的《京津冀及周邊地區(qū)2017 年大氣污染防治工作方案》��,以及 2018 年 1 月 1 日起施行的《中華人民共和國環(huán)境保護稅法》 等,都對原料藥產(chǎn)業(yè)環(huán)保進行了強力監(jiān)管[10]。原料藥的生產(chǎn)因受環(huán)保��、防爆防污染等諸多因素影響���,申請人不具備或不完全具備商業(yè)化生產(chǎn)的條件��,往往需要委托生產(chǎn)��,或?qū)⒉糠稚a(chǎn)工序委托給其他生產(chǎn)企業(yè)甚至化工企業(yè)�。個別申請人將制劑產(chǎn)品不同工序分別委托不同企業(yè)進行生產(chǎn),或受托生產(chǎn)企業(yè)將受托品種部分生產(chǎn)工序再次委托其他企業(yè)生產(chǎn)的情況也實際存在�����,如將制劑的前段生產(chǎn)工藝(固體分散體����、微粉化)等委托給其他企業(yè)進行生產(chǎn)等�。
2.3 新建車間或生產(chǎn)線���,藥品生產(chǎn)質(zhì)量管理規(guī)范(GMP)符合性情況有待確認
化學藥創(chuàng)新藥的生產(chǎn)����,尤其是原料藥的生產(chǎn)大多在新建車間或生產(chǎn)線進行,雖在注冊申報前取得了相應的藥品生產(chǎn)許可證,但在進行注冊現(xiàn)場核查前尚未進行 GMP 符合性檢查,是否具備上市商業(yè)化生產(chǎn)條件需在注冊現(xiàn)場核查時予以重點關注并確認。
三�����、化學藥創(chuàng)新藥注冊核查的主要問題
基于化學藥創(chuàng)新藥在注冊現(xiàn)場核查中的特點����,本研究主要針對這些特殊性,梳理了化學藥創(chuàng)新藥在注冊現(xiàn)場核查發(fā)現(xiàn)的主要問題。
3.1藥物研發(fā)質(zhì)量管理體系不完善
3.1.1 研究不充分����,注冊過程中持續(xù)變更
部分化學藥創(chuàng)新藥申請人急于提交上市申請����,還未進行充分研究�����,確認與驗證亦不充分就提交了注冊申請,在現(xiàn)場核查時發(fā)現(xiàn)部分創(chuàng)新藥的生產(chǎn)操作�、質(zhì)量標準等在注冊申報后進行了變更��,個別原料藥所用起始物料的來源也發(fā)生了變更;部分化學藥創(chuàng)新藥存在工藝驗證不充分�����、設備驗證確認不足的問題�。例如:某申請人對某片制粒干混工序和總混工序的生產(chǎn)操作進行了變更,部分物料的質(zhì)量標準也進行了變更�����,與申報資料不一致��;某注射劑未對配制溶液的均一性進行驗證�;某多規(guī)格片劑未對不同規(guī)格包裝工序進行驗證�;某原料藥儲存溫度為 2 ℃ ~ 8 ℃ ,未進行運輸確認����;配液系統(tǒng)進行設備確認時�,未進行攪拌速率及溫度的確認等�。
建議申請人遵循研發(fā)規(guī)律,強化質(zhì)量管理�,按照《藥品注冊管理辦法》第三十四條的規(guī)定��,在完成支持藥品上市注冊的藥學、藥理毒理學和藥物臨床試驗等研究��、確定質(zhì)量標準��、完成商業(yè)規(guī)模生產(chǎn)工藝驗證���,并做好接受藥品注冊核查檢驗的準備后�����,提出藥品上市許可申請�����,按照申報資料要求提交相關研究資料����。一旦發(fā)生重大變更����,申請人要向 CDE 進行申報,加強變更管理和研究。
3.1.2 不重視注冊申報品種的共線生產(chǎn)風險評估
由于創(chuàng)新藥研發(fā)周期長����、投入成本高��,在注冊申報時申請人可能不具備生產(chǎn)條件��,委托生產(chǎn)的情況較為突出,分階段委托生產(chǎn)組織模式共線品種多����、生產(chǎn)線和設備更復雜�����。在委托生產(chǎn)中,受托企業(yè)同時承擔其他品種的生產(chǎn)�����,無法做到對新增品種進行共線生產(chǎn)可行性評估����,或共線生產(chǎn)風險評估流于形式�����。特別是在臨床試驗用藥品生產(chǎn)階段�����,由于創(chuàng)新藥的藥理毒理數(shù)據(jù)不充分,未進行共線評估即進行共線生產(chǎn),清潔效果確認不充分等都是注冊現(xiàn)場核查中較為典型的案例�����。個別企業(yè)將細胞毒性產(chǎn)品與非細胞毒性產(chǎn)品共線生產(chǎn)�,未充分評估共線生產(chǎn)帶來的風險。共線生產(chǎn)的風險評估過于簡單,沒有綜合考慮藥品的特性與預定用途等因素,未全面分析殘留物具體的溶解性�、毒理數(shù)據(jù)等數(shù)據(jù)進行評估��。在污染與交叉污染控制方面存在不足,未能對產(chǎn)品生產(chǎn)全過程進行有效控制。
建議申請人將共線生產(chǎn)策略納入質(zhì)量協(xié)議中����,共線生產(chǎn)風險評估應當充分考慮品種在不同生產(chǎn)階段的共線生產(chǎn)情況�����,評估應更加充分,并采取足夠的措施降低交叉污染風險;應充分了解共線產(chǎn)品特性(藥理毒理、安全性)及清潔難易程度���,制定可操作性強、科學����、合理的清潔程序并應進行驗證����。必要時��,可根據(jù)風險評估來決定是否使用專用或獨立設施設備。
3.1.3技術轉(zhuǎn)移研究不充分
部分企業(yè)對產(chǎn)品技術轉(zhuǎn)移理解不夠�,研究不充分��。生產(chǎn)工藝轉(zhuǎn)移不充分或未成功即開展商業(yè)規(guī)模生產(chǎn)工藝驗證,工藝驗證中出現(xiàn)偏差無法找到根本原因�����。檢測方法轉(zhuǎn)移不充分�,造成對檢驗結(jié)果超標(out of specification,OOS)調(diào)查困難���,復測頻率、復測數(shù)據(jù)增加���,企業(yè)未采取措施對產(chǎn)生的復測數(shù)據(jù)進行有效的管理,評估對研究結(jié)果的影響[11]�。
創(chuàng)新藥研發(fā)不同于仿制藥�,可以借鑒參比制劑的經(jīng)驗進行比對研究��。新藥研發(fā)過程中的質(zhì)量標準的制定����,往往需要對檢測方法進行開發(fā),需要對新建立的方法進行方法學驗證�,但現(xiàn)場核查時發(fā)現(xiàn)�,企業(yè)在技術轉(zhuǎn)移過程中對檢測方法的轉(zhuǎn)移做得不充分�,以至于在商業(yè)規(guī)模生產(chǎn)工藝驗證階段出現(xiàn) OOS 調(diào)查不充分,無法證明是方法本身的問題還是產(chǎn)品質(zhì)量的問題�����。
從藥品研制到生產(chǎn)階段的技術轉(zhuǎn)移是一個系統(tǒng)工程����,其目的是將在研制過程中所獲取的產(chǎn)品知識和經(jīng)驗轉(zhuǎn)移給生產(chǎn)企業(yè)。建議申請人參照美國注射劑協(xié)會( Parenteral Drug Association,PDA)及世界衛(wèi)生組織等發(fā)布的關于技術轉(zhuǎn)移的相關指南要求開展化學藥創(chuàng)新藥的技術轉(zhuǎn)移工作���,加強技術轉(zhuǎn)移管理��。對生產(chǎn)工藝����、檢測方法等的轉(zhuǎn)移制定詳細的轉(zhuǎn)移方案��,在轉(zhuǎn)移過程中出現(xiàn)的偏差�、OOS 等建立相應的管理制度��,做好研究工作的銜接�。
以上問題的產(chǎn)生均反映出申請人在藥學研制環(huán)節(jié)質(zhì)量管理的薄弱���。目前針對藥學研制環(huán)節(jié)的規(guī)范性要求尚無明確的技術標準���,行業(yè)內(nèi)藥學研制過程中質(zhì)量管理水平參差不齊�、尺度不一,給藥品審評及核查結(jié)論的判定帶來一定影響�����。建議組織制定針對藥學研究機構(gòu)的質(zhì)量管理規(guī)范以及相關要求���,以提高藥學研制環(huán)節(jié)的規(guī)范性��,保證核查結(jié)果的判定有規(guī)可依��。
3.2藥品注冊申請人與受托單位之間的職責不清
按照《藥品注冊管理辦法》,藥品注冊申請人取得藥品注冊證書后為藥品上市許可持有人(MAH)�����。隨著 2019 年《中華人民共和國藥品管理法》的發(fā)布實施����,藥品 MAH 制度正式以法律的形式在中國建立����。MAH 制度下生產(chǎn)許可和上市許可實現(xiàn)分離,可更好地調(diào)動和發(fā)揮市場資源配置的主動性和靈活性,鼓勵創(chuàng)新[12]�?;瘜W藥創(chuàng)新藥的上市許可就充分利用了 MAH 制度的紅利���,在提交上市申請時進行分段��、多場地委托生產(chǎn)���。中國現(xiàn)行 MAH 制度中對于MAH 的資質(zhì)�����,以及對藥品研制、生產(chǎn)、經(jīng)營與使用環(huán)節(jié)的安全、有效及質(zhì)量可控負責作出了要求[13- 15]。但在成為 MAH 之前���,處于藥品注冊申請人的角色中,注冊現(xiàn)場核查時仍然發(fā)現(xiàn)申請人與受托單位之間分段生產(chǎn)的職責劃分不明����,對委托生產(chǎn)的原料藥或制劑的變更�����、偏差管理不完善���,對委托單位的質(zhì)量審計不充分等問題[16]�。
建議申請人通過細化質(zhì)量協(xié)議的方式與受托企業(yè)約定質(zhì)量責任�,明確對委托生產(chǎn)過程中的偏差�、變更、OOS、放行等進行審核的職責��,加強對受托企業(yè)的現(xiàn)場審計��,有效實現(xiàn)對受托企業(yè)質(zhì)量管理體系運行情況的把控�����,落實主體責任,確保受托藥品的質(zhì)量。從核查角度,建議明確針對注冊申請人分段委托生產(chǎn)的核查要點,在現(xiàn)場核查時予以重點關注�。從監(jiān)管角度�����,建議明確生產(chǎn)許可中對于分段、多場地委托生產(chǎn)的規(guī)定,統(tǒng)一境內(nèi)外分段生產(chǎn)的要求,加強對多場地和委托生產(chǎn)的監(jiān)管�。
3.3 GMP 符合性的問題突出
按照《藥品注冊管理辦法》第四十七條���,對于創(chuàng)新藥���、改良型新藥以及生物制品等���,應當進行藥品注冊生產(chǎn)現(xiàn)場核查和上市前藥品生產(chǎn)質(zhì)量管理規(guī)范檢查(簡稱“上市前藥品 GMP 檢查”)�。在對近 3 年114 個化學藥創(chuàng)新藥的注冊核查任務統(tǒng)計過程中發(fā)現(xiàn)�����,僅有 26 個(占比 22. 8% )在注冊核查時同步開展了藥品 GMP 符合性檢查��,發(fā)現(xiàn)存在主要缺陷的有10 個。對于此種情況,各省檢查機構(gòu)開展 GMP 符合性檢查的規(guī)則和尺度尚未統(tǒng)一。同時 CDE 對于注冊現(xiàn)場核查通常僅要求開展靜態(tài)核查�����,GMP 符合性檢查不開展或僅橋接判斷��,可能存在潛在風險。
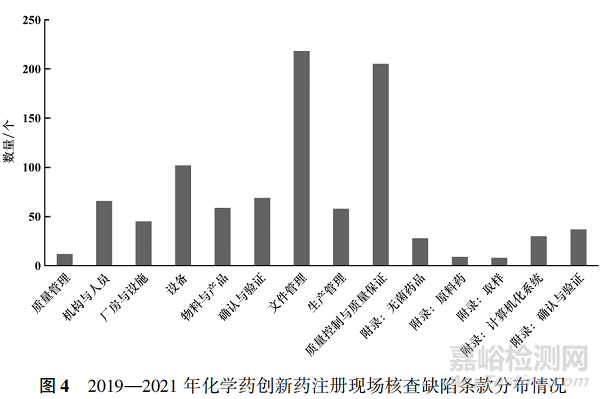
圖 4 為注冊現(xiàn)場核查中發(fā)現(xiàn)的 GMP 缺陷����,共計946 條���,其中文件管理和質(zhì)量控制與質(zhì)量保證方面缺陷占比最高���,分別為 23% 和 21. 6% ����;其次為設備����、確認與驗證方面的缺陷占比較高,分別為 10. 8% 和8. 8% �。雖然注冊現(xiàn)場核查過程中也會關注藥品生產(chǎn)企業(yè) GMP 的情況���,但藥品注冊現(xiàn)場核查不是全體系的 GMP 檢查��。
為了確保上市產(chǎn)品生產(chǎn)的持續(xù)合規(guī),建議創(chuàng)新藥申請人主動向當?shù)厥【痔岢錾鲜星?GMP 檢查的申請�,在藥品注冊生產(chǎn)現(xiàn)場檢查時同步進行藥品GMP 符合性檢查�。對在注冊核查中發(fā)現(xiàn)的 GMP 缺陷在產(chǎn)品上市前應進行全面整改���,并將整改情況報所在地省局�����。建議屬地監(jiān)管機構(gòu)加強創(chuàng)新藥上市后GMP符合性監(jiān)督檢查與抽樣檢驗����,強化監(jiān)管力度��。
四��、結(jié)語
研發(fā)環(huán)節(jié)是創(chuàng)新藥生命周期的源頭,藥品的基本屬性取決于研發(fā)質(zhì)量�,因而研發(fā)環(huán)節(jié)的監(jiān)管也是國家藥品生命周期監(jiān)管的起點�����,藥品注冊申請人應首要履行好主體責任。質(zhì)量管理體系不完善����、與受托機構(gòu)之間的職責劃分不清以及存在 GMP 符合性問題等都可能影響創(chuàng)新藥的安全��、有效和質(zhì)量可控等成藥性指標?!吨腥A人民共和國藥品管理法》 (主席令第31號) 第六條規(guī)定了 MAH 的法律責任����,即對藥品研制�、生產(chǎn)、經(jīng)營以及使用全過程中的安全性�����、有效性和質(zhì)量可控性負責����。藥品注冊申請人取得藥品注冊證后即為 MAH,應當按照我國法律法規(guī)�����,落實主體責任�,明確與委托單位之間的職責,對質(zhì)量體系管理進一步加強����。
作者|葉笑(CFDI) 董江萍(國際交流中心)






