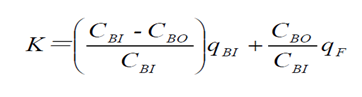剛剛,國家藥品監督管理局醫療器械技術審評中心發布《一次性使用血液透析器臨床評價注冊審查指導原則(征求意見稿)》,內容如下:
一次性使用血液透析器臨床評價注冊審查指導原則
(征求意見稿)
本指導原則旨在指導注冊申請人對一次性使用血液透析器注冊開展臨床評價,同時也為技術審評部門審評一次性使用血液透析器臨床評價資料提供參考。
本指導原則是對一次性使用血液透析器臨床評價的一般要求,注冊申請人需依據產品的具體特性確定其中內容是否合適。若不適用,需具體闡述理由及相應的科學依據,并依據產品的具體特性對注冊申報資料的內容進行充實和細化。
本指導原則是對注冊申請人和技術審評人員的指導性文件,但不包括審評審批所涉及的行政事項,亦不作為法規強制執行,需在遵循相關法規的前提下使用本指導原則。如果有能夠滿足相關法規要求的其他方法,也可以采用,但是需要提供詳細的研究資料和驗證資料。
本指導原則是在現行法規和標準體系以及當前認知水平下制定,隨著法規和標準的不斷完善,以及科學技術的不斷發展,相關內容也將適時進行調整。
一、適用范圍
本指導原則適用于一次性使用血液透析器,包括低通量透析器和高通量透析器,在醫療機構用于急、慢性成人腎功能衰竭患者,進行常規的血液透析治療。該產品分類編碼為10-04-01,管理類別為三類。
二、臨床試驗基本要求
一次性使用血液透析器通常需通過臨床試驗路徑進行臨床評價,當注冊申請人通過臨床試驗開展血液透析器的臨床評價時,可參考本章要求開展臨床試驗。如注冊申請人選擇同品種路徑進行臨床評價,需滿足本指導原則第三部分的相關要求,按照本導則第三部分內容提供相應資料。
(一)臨床試驗目的
臨床試驗需設定明確、具體的試驗目的。申請人可綜合分析試驗器械特征、非臨床研究情況、已在中國境內上市(下文簡稱已上市)同類產品的臨床數據等因素,設定臨床試驗目的。臨床試驗目的決定了臨床試驗各設計要素,包括主要評價指標、試驗設計類型、對照試驗的比較類型等,進而影響臨床試驗樣本量。
(二)臨床試驗設計類型
建議開展多中心臨床試驗,臨床試驗類型為前瞻性、隨機對照。對照組一般選擇透析參數和膜材料相似的透析器。
(三)入排標準
試驗對象需具有代表性,對入選標準和排除標準需有詳細說明。試驗對象原則上需為開展維持期血液透析的慢性腎衰竭成年患者,標明年齡、性別、原發病、特殊要求等。排除標準通常包括試驗對象伴有嚴重貧血,感染,腫瘤,活動出血,嚴重心、肝、肺臟疾病,精神異常或病情不穩定等患者等,或有其他不適合試驗的情況,如產品有風險、對患者有傷害或影響療效。
(四)評價指標
1.主要評價指標
低通量血液透析器主要評價指標包括肌酐清除率與尿素氮清除率,對于高通量血液透析器還包括β2微球蛋白(β2-MG)下降率。若注冊申請人通過增大纖維膜的標稱孔徑等宣稱擴大物質清除范圍,除以上主要評價指標外,還需根據產品自身特點增加合適的主要評價指標并論述其選擇的合理性,如考慮以λ游離輕鏈為代表的中分子物質的清除率作為主要評價指標之一。此時,需同時評價患者體內白蛋白的丟失情況,以白蛋白下降為主要評價指標,明確隨訪時間以及確定依據。
透析溶質清除率需平穩透析60min后,在固定工作狀態下的血流量和透析液流量(通常設置超濾率0或者10mL/min),同時從透析器動靜脈端抽血,檢測肌酐、尿素氮、β2-MG或λ游離輕鏈等并計算清除率。通常按照如下公式進行計算:
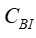 和
和 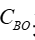 采用的濃度單位相同; 是指血液透析器血液入口的溶液濃度; 是指血液透析器血液出口的溶液濃度;
采用的濃度單位相同; 是指血液透析器血液入口的溶液濃度; 是指血液透析器血液出口的溶液濃度; 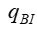 是指產品入口端的血液流速;
是指產品入口端的血液流速; 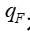 是指濾過液流速(超濾率)。
是指濾過液流速(超濾率)。
2.次要評價指標
2.1肌酐下降率、尿素氮下降率、以及適用時的代表性中分子物質的下降率。測定透析溶質下降率時,在透析開始和結束時抽血,停止超濾,血流量先減至100mL/min后,停泵立刻從患者體內抽血,透析溶質下降率=1-(透析后血濃度/透析前血濃度)%。
注:測定下降率時,在透析開始和結束時抽血,停止超濾,血流量先減至100mL/min后,停泵立刻從患者體內抽血。
2.2超濾率及超濾系數:超濾率(mL/h)=總除水量(mL)/治療時間(h)。
2.3 C反應蛋白、透析開始時和透析15min后血氣、透析開始后15min白細胞及血小板下降率等。
2.4血常規、肝腎功能、血電解質(包括血鉀、血鈣、血磷、HCO3-或CO2CP等)、白蛋白、球蛋白、C-反應蛋白。生命體征:血壓、脈搏、體溫、心率;患者一般狀態:患者自我感覺、首次使用綜合癥及體征變化;與使用透析器相關的數據變化;與透析器功能相關的實驗室參數。
2.5安全性指標:不良事件發生情況及嚴重不良事件發生情況,明確事件的具體信息如患者信息、發生時間、原因、后果、處理措施、患者轉歸及與器械的相關性等。
(五)并發癥及處理預案(預計本試驗中可能出現的并發癥及如何處理)
包括透析中低血壓、肌肉痙攣、惡心和嘔吐、頭痛、胸痛和背痛、皮膚瘙癢、失衡綜合征、透析器反應(臨床分為A型和B型反應)、心律失常、溶血、空氣栓塞、發熱、透析器破膜、體外循環凝血等。
(六)臨床試驗樣本量
根據對照用血液透析器的相應指標的循證醫學相關資料做出檢驗假設并計算樣本量。同時,樣本量的確定與選擇的假設檢驗類型(優效、非劣效、等效性檢驗)及I、II類錯誤和具有臨床意義的界值(療效差)有關,同時還需考慮預計排除及臨床失訪的病例數。
臨床試驗樣本量的確定需符合臨床試驗的目的和統計學要求,并且完成所有訪視的受試者不應少于臨床試驗方案中規定的最低樣本量。為保證各主要評價指標的檢驗功效均不低于80%,臨床試驗選擇計算例數最多的主要評價指標的樣本含量為臨床試驗樣本量。臨床試驗如為非劣效設計,非劣效界值不大于主要評價指標平均水平的10%。若申報產品包含不同膜面積產品,建議注冊申請人在1.5m2以及1.5m2-2.0m2區間內分別選擇代表性產品進行樣本量估計并開展臨床試驗。
三、同品種臨床評價基本要求
以下兩種情形下,血液透析器可通過同品種路徑開展臨床評價,一是申報產品使用的透析膜已在中國境內獲準上市,注冊申請人可選擇具有相同透析膜的透析器作為同品種醫療器械;或二是申報產品的前代產品已在中國境內獲準上市,申報產品是對前代產品透析膜壁厚及紋理/波形進行改變,注冊申請人可選擇前代產品作為同品種醫療器械。此時,血液透析器同品種臨床評價相關要求如下:
(一)適用范圍及臨床使用相關信息的對比
申報產品與同品種醫療器械需有相同的適用范圍,需比對的信息包括適用范圍、適應證、適用人群及使用時間等。
(二)技術特征的對比
1. 結構組成
注冊申請人需對比申報產品與同品種醫療器械的結構組成,關注透析膜材料和設計特征的對比,其包括但不限于透析膜材料、透析膜面積大小、纖維數量、有效纖維長度、纖維內徑、纖維壁厚、纖維膜標稱孔徑大小及分布范圍、孔隙率等。
2.性能指標
申報產品與同品種醫療器械需在同等的檢測條件下進行性能檢測,若申報產品包括不同膜面積,按照膜面積<1.5m2,1.5m2-2.0m2分別進行性能測試。透析器性能參數的比對建議重點考慮以下內容(包括但不限于):
2.1.清除率:常用尿素、肌酐、磷酸鹽、維生素B12的清除率評價透析器的濾除性能,對于高通量透析器,還需比對申報產品與同品種產品β2-MG的清除率。若注冊申請人通過增大纖維膜的標稱孔徑等擴大物質清除范圍,還需根據產品自身特點增加合適物質的清除率并論述其其選擇的合理性,如考慮以λ游離輕鏈為代表的中分子物質的清除率。
測定清除率需覆蓋注冊申請人申報的血液流速和透析液流速范圍。相關物質的清除率測定可選擇透析液流速的最低和最高點,并分別對應注冊申請人申報的血液流速的最低流速、每增加100mL/min的血液流速,直至最高血液流速。
2.2.超濾率與超濾系數:超濾率可評價血液透析器對水的清除能力。測定超濾率需覆蓋注冊申請人申請的跨膜壓(TMP)和血液流速的范圍。將測定的多個超濾率及其對應的TMP進行線性回歸,以計算超濾系數。如高通量透析器,可在600-1800mL/h超濾率范圍內,測定至少4個數據點的超濾率并記錄該超濾率下的TMP,通過超濾率與TMP的斜率來計算其超濾系數。
2.3.壓力降:測定壓力降需覆蓋注冊申請人申請的血液流速及透析液流速范圍。建議在血液流速為100mL/min、200mL/min、300mL/min、400mL/min時測量血液側的壓力降;在透析液流速為500mL/min、600mL/min、700mL/min、800 mL/min時測量透析液側的壓力降。當最高血液流速和最高透析液流速與上述建議值不同時,還需分別測量最高血液流速和最高透析液流速下的壓力降。
2.4. 篩選系數:高通量透析器還需測定篩選系數。建議在血液流速為 200mL/min、300mL/min、400mL/min、500mL/min以及最大流速(當與上述流速不同時)時測量白蛋白、菊粉、肌紅蛋白或β2-MG的篩選系數。
(三)差異性部分的安全有效性證據
1. 申報產品為新注冊產品,其使用的透析膜已在境內獲準上市,注冊申請人選擇具有相同透析膜的透析器作為同品種醫療器械時,注冊申請人需按照上述要求進行結構組成、透析膜材料和設計特征以及相關性能指標的對比,在充分識別申報產品與同品種醫療器械相同性和差異性的基礎上,論證二者的等同性。如申報產品的設計影響血液流動模式,或是最大血液流速與同品種醫療器械存在明顯差異,申請人需提供最大血液流速下機械性溶血測試數據,以驗證該設計不會引起紅細胞的過度溶解。建議在最大血液流速下進行測試。
2. 申報產品的前代產品已在中國境內獲準上市,申報產品是對前代產品透析膜的壁厚及紋理/波形進行改變,注冊申請人選擇前代產品作為同品種醫療器械,在結構組成、透析膜材料和設計特征以及相關性能指標對比的基礎上,還需針對差異部分開展臨床試驗,以確認血液透析器的臨床性能。
2.1總體要求:通過12名受試者接受總計36次治療的數據,驗證血液透析器的臨床性能。如申報產品包括不同膜面積,建議在膜面積<1.5m2以及1.5m2-2.0m2范圍內各自選擇代表性產品,分別開展臨床試驗,
2.2評價指標
2.1.1超濾率和超濾系數:每名受試者測定4個數據點的超濾率,通過線性回歸計算超濾率與其對應的TMP的斜率,獲得體內超濾系數。例如對于高通量透析器,在預先設定的時間點,超濾率分別為600mL/h、1000mL/h、1400mL/h、1800mL/h,5分鐘之后記錄實際TMP。通過12例受試者的超濾系數,反映透析器體內的超濾系數,以體內外外超濾系數的百分比評價體內外超濾系數的一致性。
2.2.2尿素氮、肌酐、磷酸鹽清除率、β2-MG下降率(高通量透析器)。
2.2.3透析開始后15min白細胞、血小板下降率,透析開始時和開始后15min的血氣分析,透析前后的紅細胞、白細胞、血小板變化情況。
2.2.4不良事件的發生情況、處理方式及處理結果,包括補體激活和血栓形成等。
2.3數據分析及結論
注冊申請人需對臨床試驗結果進行分析,評價產品的臨床性能。
(四)同品種產品的臨床數據
建議參考《醫療器械臨床評價技術指導原則》要求,提交同品種產品的臨床數據;臨床試驗或臨床使用獲得的數據(以下簡稱臨床數據)可來自中國境內和/或境外公開發表的科學文獻和合法獲得的相應數據,包括臨床試驗數據、臨床經驗數據、臨床文獻數據等。臨床文獻數據的收集應保證查準、查全,具有可重復性;臨床經驗數據收集應包括對已完成的臨床研究、不良事件、與臨床風險相關的糾正措施等數據的收集。注冊申請人可依據產品的具體情形選擇合適的數據來源和收集方法。
同品種醫療器械臨床數據需在國內現行血液透析操作規范的條件下(特別是血液流速與治療次數)收集。
四、參考文獻
[1]《醫療器械臨床評價技術指導原則》(2021年第73號通告)[Z].
[2]《決策是否開展醫療器械臨床試驗技術指導原則》(2021年第73號通告)[Z].
[3]《醫療器械臨床試驗設計指導原則》(2018年第6號通告)[Z].[4]《醫療器械臨床評價等同性論證技術指導原則》(2021年第73號通告)[Z].[5]《醫療器械注冊申報臨床評價報告技術指導原則》(2021年第73號通告)[Z].
[4]《一次性使用透析器產品注冊技術審查指導原則》(2013年第3號通告)[Z].