摘 要
長效注射劑已逐漸用于抗精神分裂癥治療�,棕櫚酸帕利哌酮長效注射劑有效控制藥物釋放,有良好的安全性、臨床治療效果和明顯的臨床優勢��,可以一定程度上提高患者依從性�。本文通過檢索相關國外審評報告和文獻資料,概述國內外上市申報的棕櫚酸帕利哌酮長效注射劑情況,對此類藥物的藥學研究進行探討,提出關注處方工藝和質量控制等研究的考慮,為其研究開發提供一定的參考��。
棕櫚酸帕利哌酮是帕利哌酮的棕櫚酸酯���,在體內水解生成帕利哌酮��。帕利哌酮為第2代抗精神病藥物��,是利培酮的活性代謝物9-羥基利培酮,為選擇性單胺能拮抗藥,對多巴胺D2受體和5-羥色胺5-HT2A受體具有拮抗作用����。精神分裂癥治療過程相對較長�����,部分患者用藥依從性差����,可能存在自行吐藥或者抗拒吞咽情況,一些特殊注射劑可以達到長效或減毒的目的等��。作為利培酮代謝產物前藥制劑��,棕櫚酸帕利哌酮長效注射劑利用納米晶體技術制備����,通過劑型的改良滿足了臨床需求�,利于維持長期治療效果、降低復發率等����,具有明顯的臨床應用價值�,也延長了產品的生命周期。
長效注射劑(LAI)可以經皮下或肌內注射等途徑給藥�����,在注射位點形成藥物儲庫發揮長效釋放作用���。LAI能提高藥物的便利性(減少給藥頻率)和治療效果(控制藥物釋放和降低藥物不良反應發生率)�。但同時技術壁壘較高,在藥學研究時對輔料、工藝技術和商業化生產能力等具有較高要求,且突破原研專利較難���,較難被仿制。
本文結合上市藥物的相關專利和文獻,針對棕櫚酸帕利哌酮長效注射劑從處方工藝研究�����、質量研究控制等方面探討本品仿制藥藥學研究可以關注的問題�。
1、棕櫚酸帕利哌酮長效注射劑的國內外上市情況
1.1 美國上市情況
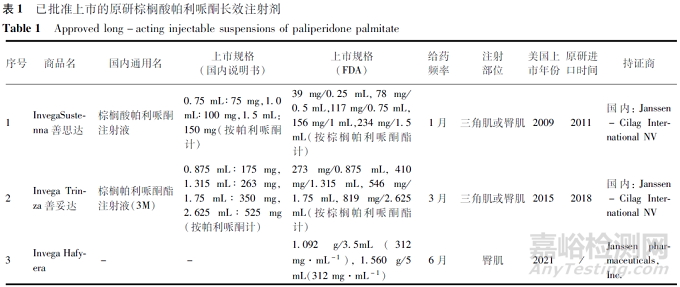
棕櫚酸帕利哌酮由楊森公司開發,目前上市原研制劑包含1月制劑(PP1M)���、3月制劑(PP3M)和6月制劑(PP6M)。PP1M(商品名Invega Sustenna,NDA022264)于2009年在美國上市��,上市5個規格����。PP3M(商品名Invega Trinza���,NDA0207946)于2015年在美國上市�����,上市4個規格�。PP6M(商品名Invega Hafyera)于2021年在美國上市���,上市2個規格(見表1)���。
美國食品藥品監督管理局(FDA)鼓勵仿制長效注射劑的研發�����,可以在一定程度上使患者更好地獲得可負擔的藥物�����。FDA發布了PP1M和PP3M的個藥指南(PSGs)以指導仿制藥研發,2022年FDA計劃修訂的個藥指南列表中包含PP1M�,將對體內研究設計做微小的修改�����。
2021年7月FDA批準了以Invega Sustenna(NDA022264)為參比制劑(RLD)的首個仿制棕櫚酸帕利哌酮注射液,仿制藥(ANDA)編號為211149�,持證商為Teva公司�����,暫未銷售�。FDA在2021年度仿制藥報告中提到此仿制藥使用了FDA開發的用于藥代動力學研究設計和生物等效性評估的模型和模擬方法。其FDA批準信中說明原研存在專利保護期(美國專利的最后到期日2031年),仿制企業在ANDA申報資料中包含了專利第Ⅳ段聲明�,截至2022年11月暫未獲得180天仿制藥市場獨占期���。FDA公布的第Ⅳ段聲明列表(更新日期2022-10-03)中也顯示目前有1家仿制藥企業針對Invega Sustenna提交了此聲明�����,“180天狀態”為“推遲”。
1.2 歐盟和日本上市情況
歐盟已上市原研PP1M(商品名Xeplion)���,規格為25、50����、75�����、100和150 mg(按帕利哌酮計)��,和上市原研PP3M(商品名Trevicta),規格為175�、263��、350、525 mg(按帕利哌酮計)���。歐洲已有多家PP1M仿制藥通過互認程序(MRP)批準上市,各國間產品名存在差異�,例如Dopelir參考成員國(RMS)為丹麥����,Paliperidone Biogaran的RMS為法國�����。日本也上市了原研PP1M(Xeplion)和PP3M(Xeplion TRI)。
1.3 中國上市申報情況
國內僅有原研產品PP1M和PP3M上市,分別上市了3個規格和4個規格(見表1)����。截至2022年12月底�����,暫無國產制劑上市,國內已有2家國產企業申報PP1M上市、1家國產企業申報PP3M。
國家藥品監督管理局藥品審評中心發布的《化學仿制藥參比制劑目錄》(截至2022年11月第57批)已公布棕櫚酸帕利哌酮長效注射劑的多個參比制劑,包括原研進口和未進口原研藥品(美國橙皮書、歐盟上市)�����,包含PP1M 13個����、PP3M 12個,目前暫未有PP6M的參比制劑公布。仿制藥企業首先可以結合品種情況選擇合適的參比制劑��。
2����、藥學研究探討
2.1 處方工藝
國外的[FDA、歐洲藥品管理局(EMA)和日本藥品和醫療器械管理局(PMDA)]審評報告和說明書顯示PP1M處方組成包括無菌原料藥、聚山梨酯20����、聚乙二醇4000���、一水檸檬酸����、無水磷酸氫二鈉���、磷酸二氫鈉一水合物����、氫氧化鈉和注射用水�,PP3M和PP6M緩沖鹽中去除了無水磷酸氫二鈉?;谒幋鷦恿W模擬��,PP3M劑量是PP1M的3.5倍,同時為了減少給藥體積�����,PP3M中原料藥濃度由PP1M的156 mg·mL-1(以棕櫚帕利哌酮酯計)增加到321 mg·mL-1�����,輔料的用量有一定的差別����。FDA說明書中PP6M還明確了各輔料的用量�,聚山梨酯20(10 mg·mL-1)、聚乙二醇4000(75 mg·mL-1)���、一水檸檬酸(7.5 mg·mL-1)����、磷酸二氫鈉一水合物(6 mg·mL-1)�、氫氧化鈉(5.4 mg·mL-1)和注射用水,較PP3M增加了注射體積����。FDA和EMA的PP3M審評報告中說明了不同規格存在過量灌裝情況����。對于特殊注射劑����,仿制藥處方原則上應與參比制劑一致����。若存在過量灌裝需要通過與參比制劑進行對比等研究提供充足的依據,比如可以進行仿制藥與參比制劑可抽取藥量的對比等�。
關于原料藥的性質對制劑的影響��,FDA和EMA的審評報告顯示,棕櫚酸帕利哌酮以晶型A穩定存在�,在較寬的pH內和水中幾乎不溶���,這種低溶解度原料藥使制備長效混懸肌內注射劑成為可能���。此長效注射劑的釋藥機制主要為在進行肌內注射后����,棕櫚酸帕利哌酮在注射部位緩慢溶解釋放����,代謝為帕利哌酮,進一步被循環系統吸收從而發揮作用�����。影響制劑體內生物等效的關鍵是注射部位棕櫚酸帕利哌酮的溶解行為�����。
根據本品的釋藥機制可知�����,原料藥的粒度和粒度分布是影響產品體內釋放和起效的關鍵因素���。EMA報告顯示PP1M生產過程中采用無菌粉碎來減小原料藥粒徑�,以達到合適的粒徑范圍,而PP3M通過控制工藝增大原料藥粒度,使藥物體內溶解和釋放速度變慢��。PP3M由于與PP1M的給藥量�����、粒徑�、給藥體積的不同����,有更慢的釋藥速度。粒徑控制方面建立了經過驗證的粒徑測定方法,能夠準確測定生產�、釋放和貯藏過程中粒徑的變化���。建立了針對不同粒徑有區分性的體外釋放的方法���,并進行了粒徑和釋放的體內外相關性研究(IVIVC)����。EMA報告顯示PP3M提供了新的體內外相關性的研究數據(因為PP3M的粒徑不在PP1M 的IVIVC已驗證的范圍內)����,其臨床研究顯示一定范圍內混懸劑粒徑的增大可以降低Cmax和延長Tmax�����。對原料藥其他的理化性質�����,如純度�����、結晶性和形態學等進行了研究,進一步評價原料藥關鍵質量屬性對制劑特性的影響。
在制劑開發中適宜的輔料也起著重要作用�����,其決定了混懸液是否具有適宜的溶液外觀�����、pH值�、黏度�����、滲透壓����、再分散性和物理化學穩定性等�����。原研專利顯示聚山梨酯20屬于潤濕劑����,能夠降低水和原料的表明張力����;聚乙二醇4000作為懸浮劑�,在保持一定的相對用量時使原料藥懸浮分散在介質中;使用無水磷酸氫二鈉和磷酸二氫鈉一水合物來做為緩沖劑調節等滲和中性的環境�,且使其中的懸浮酯不易于絮凝�����。EMA報告顯示氫氧化鈉做pH調節劑。同時由于考慮給藥方便性���,需混懸液保持合適的黏度,使短時間內在較細的針頭中較易通過�����。EMA的PP3M可重懸浮性研究采用3批已在室溫儲存了9至11個月的驗證批次進行��,并且在說明書中詳細說明用前振搖的方法�����。
工藝開發方面,EMA審評報告中顯示制備過程主要包括將無菌原料藥分散在無菌介質中��,混懸液進行濕法研磨達到目標粒徑����,用注射用水稀釋后進行無菌灌裝。擬定的關鍵步驟為除菌過濾�����、研磨和灌裝��,同時制定了一系列的過程控制措施。對于無菌生產工藝,仿制藥處方工藝開發中應充分考慮各生產步驟的無菌控制���。在工藝描述時如果能夠充分描述各步驟�,特別是除菌過濾�����、研磨和灌裝的工藝參數更有利于理解工藝過程����,例如除菌過濾可以明確過濾器材質��、型號等信息�����,并監測其過濾前后完整性和過濾壓力等參數,比較容易遺漏除菌過濾前料液的微生物負載情況���。研磨步驟可以關注研磨機轉速、研磨時間��、研磨介質使用情況(如介質裝量)等參數����。進行無菌工藝驗證注意可以包含培養基灌裝試驗,直接接觸無菌物料和產品的設備����、部件�、容器密封性系統的除菌除熱原驗證等��。目前濕法研磨技術較為成熟并得到了廣泛使用����,市場上也有不同品牌的納米研磨機可供研究和生產使用��。關于批量問題,針對此類特殊注射劑注冊批和商業批的批量原則上應保持一致。
包材選擇方面�,原研制劑內包裝均采用透明的環烯共聚物(COC)預灌封注射器(PFS)(連接帶膠塞推桿�����、護帽),配有逆止器和安全針頭。EMA審評報告中顯示申請人已對包材的相容性和密封性均進行了詳細研究。使用中穩定性研究則對給藥過程中的功能性進行了研究��,包括針頭與注射器對拆離力矩(PRF)���、軸向分離力(PTF)��。仿制藥是否選擇與參比制劑相同的包材����,需對選擇的包材提供充分的依據��,也可以關注粒度與針頭的匹配性��。
2.2 質量研究與質量標準
長效注射劑除需要滿足常規注射劑的基本要求外,通常會結合制劑特點和自身釋藥特性等有一些特殊研究內容。EMA的PP3M審評報告中在質量控制方面對性狀/可重懸浮性/可注射性���、鑒別(HPLC、IR)、pH(6.5~7.5)��、有關物質�、含量均勻度、不溶性微粒、體外溶出����、粒度分布����、無菌�、細菌內毒素和含量進行了研究����。
藥品審評中心發布的《化學藥品注射劑(特殊注射劑)仿制藥質量和療效一致性評價技術要求》中針對特殊注射劑的質量屬性方面有一些考慮?����?疾斓年P鍵質量屬性可能包括但不限于以下內容:理化性質(如性狀�、黏度��、滲透壓摩爾濃度、pH值/酸堿度等)��,Zeta電位�,粒子形態,粒徑及分布��,體外溶出/釋放行為���,藥物晶型和結晶形態�。仿制藥可以參考此技術要求結合品種特性對部分質量屬性進行研究,原則上應提供至少3個批次參比制劑樣品的質量對比考察數據。例如針對此長效制劑,黏度不但會影響穩定性(過低的黏度會加速藥物晶體的沉淀),對通針性和給藥器內藥物殘留等均有影響。
溶出度作為關鍵質量控制指標,國外審評報告顯示原研制劑進行了IVIVC研究���。目前未查詢到此類長效注射劑在藥典中或者通用的體外溶出方法,而且建立IVIVC也面臨一些挑戰。仿制制劑可以在生物等效性試驗前在體外考察自制樣品與參比制劑溶出相似情況���,一定程度上降低生物等效性不等效風險(但有時體內外相關性并不好);標準中增加此控制可以評價自制商業批樣品與關鍵批次(例如臨床試驗批次)的質量批間一致性等。通常采用多點法制定研究限度�����,其各階段時間點選取和限度的設置可以考慮結合仿制制劑自身特性和多批關鍵批次(例如臨床批次)的質量研究和穩定性數據等擬定����,提供充分合理的依據,而非僅參考原研制劑擬定的標準�。FDA溶出度數據庫中提供了可供參考的PP1M和PP3M的溶出方法:槳法�����,轉速50 r·min-1��,溶出介質為含0.489%吐溫20的0.001 mol·L-1的HCl�,溶出介質體積為900 mL��,PP1M的取樣時間為1.5��、5、8����、10����、15��、20��、30和45 min,PP3M的取樣時間為5���、30、60����、90����、120�����、180�����、240����、300和360 min。
2.3 穩定性研究
EMA的PP3M審評報告中指出穩定性研究的考察指標包括性狀/可重懸浮性/可注射性、粒度分布、體外溶出、pH、有關物質�����、無菌�、細菌內毒素和含量,另外增加了醛含量���、質量損失和容器密封性指標��。采用正置和倒置不同的放置方式,穩定性考察條件包括了長期條件(25 ℃/40% RH��、30 ℃/35% RH)����、中間條件(30 ℃/75%RH)和加速條件(40 ℃/不超過25%RH)。在50 ℃強力試驗條件下時觀察到粒徑的明顯上升且體外釋放降低�,這是由于奧斯特瓦爾德熟化現象��。這種粒徑增長現象也可以在加速6個月(40 ℃/25%RH)和長期12個月(30 ℃)的情況下觀察到。在凍融或者冷凍情況下�,可以觀察到粒徑在初期明顯下降��。原研制劑說明書中明確了儲存溫度且說明請勿冷凍保存。PP3M最小灌裝量(0.875 mL)樣品在6月(40 ℃/25%RH)有輕微失水情況����,可能是由于半透的注射器造成�����。仿制藥在進行穩定性研究時可以結合自身包材選擇情況等按照ICH指導原則擬定合理的考察條件���。
混懸型注射劑會發生奧斯特瓦爾德熟化現象�����,即小粒子溶解并分布在大粒子表面���,小粒子逐漸減少和粒度的增長�����,導致混懸劑比表面積減少,并進一步導致溶出度的下降(藥物的溶解度與顆粒粒度大小呈反比)。而高溫一般增加了藥物晶體在懸浮液中的溶解度���,更易加劇此現象的產生。混懸型注射劑開發時通常會從優化制劑組成(如降低共溶劑或表面活性劑的水平、增加黏度�����、縮窄粒度分等)和控制產品的儲存條件等方面��,減少奧斯特瓦爾德熟化現象的發生�����。仿制藥處方工藝篩選過程也可以進行適當的穩定性考察,進一步確認原料藥粒徑分布選擇范圍�、輔料型號等選擇的合理性�����。
3、結 語
此納米晶混懸注射液對難溶性藥物有高藥物負荷的優點����,且可以使用小針頭注射,使藥物局部集中���,可最小化組織損傷的風險;其可以減少復發和用于治療的維持階段等進而提高療效和治療依從性���,因為諸多優勢,抗精神病領域基于納米晶體技術的長效注射液獲得較大市場和巨大的商業價值����。但是工業界也面臨諸多挑戰�,如生產問題(擴大生產�、無菌保障水平、可注射性等)��、IVIVC建立(長效注射劑比口服制劑有更差的體內外相關性����,獲得A級相關更為困難)等。目前國內暫無國產櫚酸帕利哌酮長效注射劑獲準上市����,其具有良好產業化前景����,值得業界開發應用和探討,以更好地造?��;颊摺?/span>