枸櫞酸西地那非是一種環磷酸鳥苷(cGMP)特異的 5 型磷酸二酯酶(PDE5)的選擇性抑制劑 ,PDE5廣泛存在于血管平滑肌、呼吸道平滑肌、血小板、胃腸道上皮細胞等,通過特異性水解細胞內第二信使 cGMP,參與多種生理、病理過程的調控,是良好的藥理學靶標。枸櫞酸西地那非是選擇性PDE5 抑制劑,通過抑制血管平滑肌上的 PDE5,提高局部 cGMP 水平,松弛、擴張血管平滑肌,廣泛用于勃起功能障礙和肺動脈高壓(PAH)的治療。
查詢美國食品藥品監督管理局(FDA)及歐盟藥品監督管理局(EMA)網站相關信息,枸櫞酸西地那非片由輝瑞公司研發,1998 年 3 月在美國作為新分子實體(Type I)獲得批準[新藥申請(NDA)號020895],商品名為 Viagra®,規格 25、50、100 mg,成為美國批準的第1個用于治療男性勃起功能障礙的口服治療藥物;2005年6月,輝瑞公司的枸櫞酸西地那非片在美國被批準用于治療肺動脈高壓(NDA號 021845),商品名 Revatio®,規格 20 mg;1998 年獲歐盟批準上市;目前該產品已在美國、英國、日本以及歐盟多個國家或地區批準上市。輝瑞公司生產的原研地產化枸櫞酸西地那非片于 2000 年 5月在中國獲批上市,商品名萬艾可®,涉及 25、50、100 mg 規格,2020 年 2 月該公司 20 mg 規格枸櫞酸西地那非片獲批進口中國。
截至 2022 年 8 月 31 日,中國已批準 30 個枸櫞酸西地那非片(含口崩片)仿制藥批準文號(含 2 個境外進口、3個原研地產化產品批準文號)、1個原研進口批準文號,涉及該品種的 4 個規格(20、25、50、100 mg);經查國家食品藥品監督管理局(NMPA)藥品審評中心化學藥品目錄集(https://www.cde.org.cn/hymlj/listpage/9cd8db3b7530c6fa0c86485e563f93c7),共收載 26 條枸櫞酸西地那非片(含口崩片)信息,含原研地產化產品3條(標記為參比制劑),23個國內批準文號通過或視同通過化學仿制藥一致性評價。經查詢藥物臨床試驗登記與信息公示平臺(http://www.chinadrugtrials.org.cn/index.html),截至 2022 年 8 月 31 日,共登記枸櫞酸西地那非片(含口崩片)生物等效試驗 50條,其中 31條登記狀態為已完成、18條為進行中、1條為主動終止。
通過梳理國內、外藥品監管機構對枸櫞酸西地那非片生物等效性試驗的要求,結合近年來該藥在中國的生物等效性試驗現狀,分析該品種生物等效性試驗的主要特征,結合生物等效性試驗審評過程中遇到的多種情況提出考慮,以期為提高仿制藥質量有所裨益。
一、體內藥動學特征
枸櫞酸西地那非片口服后吸收迅速,絕對生物利用度為 41%(25%~63%)。其藥動學參數在推薦劑量范圍內與劑量成比例。消除以肝臟代謝為主[細胞色素P450同功酶3A4(CYP4503A4)途徑],生成有活性的代謝產物 ,其性質與西地那非近似。CYP4503A4的強效抑制劑(如紅霉素、酮康唑、伊曲康唑)以及 CYP450 的非特異性抑制物如西咪替丁與西地那非合用時,可能會導致西地那非血漿水平升高。西地那非及其代謝產物的消除半衰期約為4 h。
1.1 吸收和分布
枸櫞酸西地那非片吸收迅速。空腹狀態下口服 30~120 min(中位值 60 min)后達到最大血藥濃度(Cmax)。在與高脂肪飲食同服時,吸收速率降低,達峰時間(tmax)平均延遲60 min,Cmax平均下降29%。但吸收程度未受顯著影響[藥時曲線下面積(AUC)下降 11%]。西地那非濃度為 3.5 nmol·L−1時對人PDE5 酶活性的體外抑制率達 50%。在人體中,單劑口服100 mg西地那非后,平均最大游離血漿西地那非濃度約為 18 ng·mL−1或 38 nmol·L−1。西地那非的平均穩態分布容積(Vss)為 105 L,說明其在組織中有分布。西地那非及其主要循環代謝產物(N-去甲基化物)均有大約96%與血漿蛋白結合。蛋白結合率與藥物總濃度無關。
1.2 代謝和排泄
西地那非主要通過肝臟的微粒體酶細胞色素P4503A4(主要途徑)和細胞色素 P4502C9(次要途徑)清除。主要循環代謝產物是西地那非的N-去甲基化物,后者將被進一步的代謝。N-去甲基代謝產物具有與西地那非相似的磷酸二酯酶(PDE)選擇性,在體外其對 PDE5 的作用強度約為西地那非的50%。此代謝產物的血漿濃度約為西地那非的40%,故西地那非的藥理作用大約有 20% 來自于其代謝產物。N-去甲基化代謝產物被進一步代謝,其終末半衰期約為4 h。西地那非的全身清除率為41 L·h−1,終末半衰期為 3~5 h。口服或靜脈給藥后,西地那非主要以代謝產物的形式從糞便中排泄(約為口服劑量的 80%),一小部分從尿中排泄(約為口服劑量的13%)。
二、藥品監管機構的生物等效性試驗要求
2.1 美國FDA技術要求
美國FDA自1980年發布第1版《具有治療等效性的已批準藥物》(通常被稱作橙皮書)以來,每年3月發行,現行版的橙皮書為第 41 版,其中明確指定了用于仿制藥藥學和生物等效性研究的參比制劑。FDA自2010年6月開始陸續公布和更新《特定藥物的生物等效性指導原則》,為中國化學仿制藥一致性評價以及仿制藥的上市申請中生物等效性研究提供了指導與參考。美國 FDA 橙皮書中 ,輝瑞公司的商品名為Viagra(® NDA 號 N20895)的枸櫞酸西地那非片(規格 25、50、100 mg)與 該 公 司 商 品 名 為Revatio(® NDA 號 021845)的枸櫞酸西地那非片(規格 20 mg)均 標 記 為 參 比 制 劑(RLD);其 中 20、100 mg 規格同時標記為生物等效性研究對照藥品(RS)。美國 FDA 枸櫞酸西地那非片生物等效性研究指導原則( 2008年5月定稿,2008年12月修訂)中對本品生物等效性研究建議為空腹及餐后雙交叉體內研究設計,采用健康男性受試者,考察單次給藥后血漿中西地那非及代謝活性產物 N-去甲基西地那非的主要藥動學參數 AUC 及 Cmax 90% 置信區間(90%CI)在80.00%~125.00%,具體要求見表1。
2.2 中國NMPA技術要求
中國于 2021 年 12 月發布《枸櫞酸西地那非口崩片生物等效性研究指導原則》,與 FDA 枸櫞酸西地那非片生物等效性研究個藥指南相比,具有以下 4 點異同:(1)研究類型方面,僅要求開展空腹生物等效性試驗;(2)給藥方法方面,參考枸櫞酸西地那非口崩片說明書給藥方法,建議采用直接服用方式開展臨床試驗,即將口腔崩解片置于舌上,待其崩解后直接吞咽,觀察并記錄口崩片在口中完全崩解的時間及口感等;(3)給藥劑量方面,考慮該品種目前僅批準 50 mg 規格,故建議采用 50 mg 單片服用的給藥方法,且不涉及其他規格的生物等效性豁免;(4)其他項目基本與 FDA 枸櫞酸西地那非片保持一致。國內現已批準1家企業的枸櫞酸西地那非口崩片(50 mg規格)。中國已有10余家企業申報的枸櫞酸西地那非片(涉及20、25、50、100 mg規格)獲批上市,其生物等效性研究主要參考FDA枸櫞酸西地那非片生物等效性研究個藥指南,結合2016年中國發布的《以藥動學參數為終點評價指標的化學藥物仿制藥人體生物等效性研究技術指導原則》的總體技術要求開展相關研究。
2.3 歐盟及日本生物等效性研究技術要求
歐盟及日本未發布枸櫞酸西地那非片生物等效性研究技術要求。EMA主要參照美國 FDA相關要求,開展本品種的生物等效性研究的技術審評,日本藥品和醫療器械署(PMDA)則通過受試制劑與參比制劑在不同介質中有分辨力的溶出曲線代替生物等效性研究批準上市。
三、枸櫞酸西地那非片在中國的生物等效性試驗評價結果分析
參考藥審中心近年來枸櫞酸西地那非片 10 余家申請人生物等效性研究申報數據、該品種通過仿制藥一致性評價信息公開數據(https://www. cde.org. cn/yzxpj/listpage/baccb6ea4350170164a8141548c32f2e)以及參考文獻,血漿中的西地那非及 N-去甲基西地那非主要藥動學參數 Cmax、AUC、tmax等的90%CI、變異系數(CV)進行匯總,形成枸櫞酸西地那非片生物等效性試驗結果分析見下文,部分數據見表2、3。

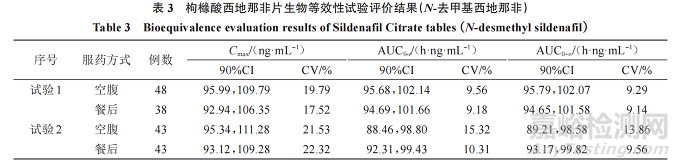
主要藥動學參數:生物等效性研究結果顯示,西地那非 Cmax最低的 90%CI下限為 80.31%,最高的90%CI 上限為 122.47%;AUC0-t最低的 90%CI 下限為87.36%,最高的90%CI上限為109.92%;AUC0-∞最低的 90%CI 下限為 87.01%,最高的 90%CI 上限為111.57%;N-去甲基西地那非最低的 90%CI 下限為81.01%,最高的 90%CI上限為 116.78%;AUC0-t最低的 90%CI 下 限 為 88.46%,最 高 的 90%CI 上 限 為114.86%;AUC0-∞最低的90%CI下限為89.21%,最高的90%CI上限為114.59%。
CV:生物等效性研究結果顯示,西地那非 Cmax的 CV 范圍為 18.17%~32.5%、AUC0-t的 CV 范圍為10.25%~17.00%、AUC0-∞ 的 CV 范 圍 為 10.03%~17.12%;N- 去 甲 基 西 地 那 非 Cmax 的 CV 范 圍 為9.78%~27.23%、AUC0-t 的 CV 范 圍 為 7.45%~15.32%、AUC0-∞的CV范圍為7.77%~15.00%。
四、枸櫞酸西地那非片生物等效性試驗審評考慮
4.1 參比制劑的選擇
截至2022年8月31日,經檢索國家藥品監督管理局已發布《化學仿制藥參比制劑目錄》第十批、第十二批、第二十三批、第二十七批、第四十批及第五十五批中發布枸櫞酸西地那非片(含口崩片)參比制劑累計11條,涉及20、25、50、100 mg 4個規 格 ,包括輝瑞公司原研進口 、原研地產化產品 、歐盟上市及美國上市的多個持證商的該產品。
參考美國 FDA 枸櫞酸西地那非片生物等效性個藥指南的相關說明,枸櫞酸西地那非片生物等效性研究需選擇對應的參比制劑開展生物等效性試驗,對于不同的參比制劑需分別提交申請,目前國內大多申請人選擇國內已批準的原研地產化產品(持證商為輝瑞制藥有限公司、商品名 Viagra®或萬艾可®)開展相關研究。由已發布的《化學仿制藥參比制劑目錄(第二十七批)》、《化學仿制藥參比制劑目錄(第五十五批)》枸櫞酸西地那非片參比制劑發布情況可知,美國橙皮書列為參比制劑的另一個枸櫞酸西地那非片原研產品(商品名 Revatio®或瑞萬托®),亦有申請人提出申請并開展仿制研究。
4.2 受試者數量、采樣點及清洗期設計
已有生物等效性研究數據可見,枸櫞酸西地那非片 AUC 及 Cmax的個體內變異為 16%~26%,如果檢驗的顯著性水平設置為 0.05,檢出受試制劑與參比制劑生物等效的研究功效為0.9,等效界限設定為 80.00%~125.00%,假設西地那非主要終點的幾何均值比為 0.95~1.07,采用 PASS 計算樣本量,則估計樣本量為 40 例,考慮約 20% 脫落風險,一般空腹及餐后入組男性健康受試者48例左右;采用單劑量、空腹與餐后,兩制劑、兩周期、兩序列、隨機、開放、自身交叉的生物等效試驗設計。
采樣點及清洗期方面,參考中國2016年發布的《以藥動學參數為終點評價指標的化學藥物仿制藥人體生物等效性研究技術指導原則》相關要求,建議每位受試者每個周期采樣12~18個采血點,采樣時間不短于 3 個末端消除半衰期,末端消除相應至少采集 3~4 個樣品以確保準確估算末端消除相斜率,AUC0-t至少覆蓋 AUC0-∞的 80%。現有研究結果表明,西地那非及其代謝物N-去甲基西地那非的終末半衰期均為3~5 h,大多研究者選擇服藥0 h(服藥前1 h內)和服藥后至24 h的18~20個采血點,清洗期一般不應短于7個半衰期,選擇7 d。
4.3 生物等效性評價
由 FDA、我國已發布的枸櫞酸西地那非片生物等效性試驗個藥指南可知,本品種的生物等效性評價要求基本一致,即受試制劑與參比制劑血漿中西地那非主要藥動學參數 Cmax、AUC0-t、AUC0-∞的幾何均值比值的 90%CI 在 80.00%~125.00%,同時提交其 代 謝 物 N- 去 甲 基 西 地 那 非 的 Cmax、AUC0-t 和AUC0-∞用于進一步支持臨床療效的可比性。
考慮受試制劑與參比制劑在生物等效性研究中藥動學參數 tmax能在一定程度上反映 2 個制劑在體內的達峰時間,一般要求申請人在提交統計分析結果是同時提交tmax非參數檢驗結果,從而進一步保障受試制劑與參比制劑的臨床等效。
五、結語
枸櫞酸西地那非片屬臨床常用藥物,該品種原研產品已在國內上市20余年,臨床療效得到廣泛認可,隨著該產品國內市場的逐年擴增,近年來成為國內外企業仿制開發的熱點品種之一;藥物臨床試驗登記與信息公示平臺顯示,截至 2022 年 8 月 31日,該品種生物等效試驗備案達 50條(含口崩片 17條)。已有生物等效性研究數據可知,枸櫞酸西地那非片體內變異總體約 26%,受試者數量一般選擇36~48例男性健康受試者開展雙交叉、兩周期空腹及餐后生物等效性試驗;枸櫞酸西地那非口崩片則建議參考已發布的《枸櫞酸西地那非口崩片生物等效性研究指導原則》僅開展空腹生物等效性試驗。同時,建議申請人提交受試制劑與參比制劑的tmax的非參數檢驗結果,進一步保障仿制藥與原研產品的臨床等效。




