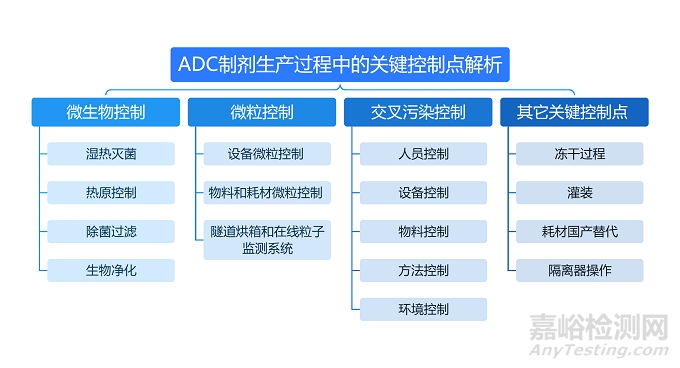
抗體偶聯藥物(ADC)兼具抗體的高特異性和細胞毒素的高抗腫瘤活性,對制劑生產也提出了更高要求。在ADC制劑生產中的經驗,ADC制劑生產過程中的關鍵控制點,從微生物控制、微粒控制、交叉污染控制以及其它關鍵點控制。
本次講詳細解析ADC制劑生產過程中的“微生物控制”。
ADC制劑生產過程中的微生物控制
ADC制劑的生產和其他大分子的生產有很多相通之處,比如對微生物的控制。微生物控制主要包含濕熱滅菌、熱原控制、除菌過濾、生物凈化四個方面:
一、濕熱滅菌
濕熱滅菌主要針對滅菌柜做器具滅菌,其中有五個注意點:
(一)滅菌柜性能:CD開發是確定滅菌柜性能的重要手段。平衡時間、BD測試和泄露率測試,這些性能測試也是考量滅菌柜質量的重要因素。
(二)針對滅菌柜的驗證:包括空載熱分布、裝載熱穿透、BI挑戰以及最大裝載/最小裝載。滅菌柜在所有驗證項目達到合格標準后才能投入生產使用。
(三)裝載策略設計:這一部分考量的是裝載的代表性以及涵蓋范圍。如果裝載發生變化,應當在前期驗證過程中盡可能地考量到,僅通過一個評估就能將變化涵蓋,從而減少驗證的工作量。裝載發生變化后,需要重新驗證的時間節點,也是設計裝載時需要重點考量的部分。
(四)生產實際過程中的運行參數:驗證通常采用Worse case參數,所以生產過程中會采用更加保守的策略。例如:驗證時,滅菌條件為121℃、30min,而在實際生產中,可以考慮用122℃、30min的參數進行生產。這樣可以保證在生產過程中,滅菌柜的溫度即使低于122度,但只要高于121度,驗證依然有效,可使風險降到最低。
(五)考慮Holding Time:通過相應的驗證來確定滅菌后的存放時限。例如一些器具滅菌以后存放多長時間。該時限的確定通常有兩個策略:一是通過相應的時限驗證來確定,二是通過模擬灌裝挑戰來確定。
濕熱滅菌的第二部分主要針對凍干機的SIP。
首先,CIP站作為為凍干機清洗提供介質的裝置,需要考量是否進行相應的SIP。純化水罐可以不進行滅菌,WFI罐則需要滅菌或至少進行蒸汽消毒處理,目的是控制微生物的限度。此外,凍干機SIP時需保證滅菌溫度和時間,以符合滅菌的要求。
其次,凍干機的SIP過程一般采用過度殺滅。由于CIP結束后,凍干機并非處于干燥狀態,導致微生物滋長,之后再進行SIP會有失敗的風險,所以CIP后不要放置太長的時間,應盡快進行SIP。在SIP中,也要關注凍干機的通氣過濾器。
第三是SIP的驗證。該驗證一般通過風險評估來確定探頭的數量以及BI的數量和點位,如對于凍干箱箱體的板層、內壁和大門,就需要通過風險評估來考慮在哪些地方布置探頭和BI。SIP的管路和過濾器則必須布置探頭和BI,以此確保管路和過濾器能夠達到無菌效果。BI在布置的過程中一定要牢固,且不能堵塞管路。如果布置不合適,可能導致SIP結束后,BI被蒸汽沖走,產生引入污染的風險,或者堵塞管路,最終造成SIP的失敗。
最后是SIP的時效性。與滅菌器柜相同,需要考察其Holding Time。該考察可以通過驗證來確定,也可以進行三批模擬灌裝來確定滅菌的時效性。通氣過濾器的滅菌次數也很關鍵,要根據過濾器的耐受次數以及實際的生產情況來綜合考察。
二、熱原控制
微生物控制第二部分是熱原控制,主要涉及隧道烘箱,可以從五個方面來解析:
(一)隧道烘箱的運行模式。在生產過程中采用的生產模式通常叫日間模式;在生產停止過程中,恰當的方式是切換成夜間模式,保持隧道烘箱的送風,防止因送風關閉造成隧道烘箱污染。
(二)冷卻段滅菌。一般每批生產前需要對冷卻段進行滅菌,保證冷卻段的無菌環境。
(三)冷卻段壓差梯度的設計。特別是對于ADC的生產,一般會使用隔離器來保證活性藥物生產的安全性,所以隧道烘箱的出口段壓差比普通隧道烘箱高。這對隧道烘箱的挑戰較大,需要防止隔離器的風灌到隧道烘箱,還要防止冷卻段的風灌到加熱段。采用雙級的壓差梯度可以避免冷卻段對高溫段的影響。
(四)隧道烘箱本身各段的壓差。要與設計保持一致。在實際運行中不能偏離設計,否則可能造成倒灌,進而影響除熱原的效果。
(五)溫度和網帶速度。這個時候要考察不同規格的西林瓶對應的除熱原溫度、網帶速度存在差別,這些參數需要通過驗證來確定。
三、除菌過濾
微生物控制需要考慮的第三部分是除菌過濾,主要是藥液的除菌過濾。
具體包含以下六個方面:
(一)濾膜材質和膜面積的選擇。目前在市面上,濾膜材質多用PVDF和PES。在實際操作中,材質的選擇要根據藥液的特性確定,通常是根據藥液的兼容性選擇。膜面積的選擇要留有緩沖余地,如果僅通過計算的值來選擇,沒有留有緩沖,可能會造成濾器的堵塞和破損,尤其要注意高粘度、高濃度、有蛋白聚集的情況,可以考慮采用預過濾和多級濾器并聯的方式來有效降低風險。
(二)時限研究。混勻后至過濾前一定要考察時限,還要考慮藥液整體的過濾時間、藥液過濾后的存放時間。
(三)影響藥液除菌過濾效能的因素。其中最主要的是藥液本身的性質,此外還有過濾工藝參數,如過濾的壓力、以及過濾器和藥液相互的作用,還有過濾量和使用周期。
(四)無菌連接。無菌連接器市面上也有很多種。此時要重點考察廠家對無菌連接器是否進行了相應驗證、驗證資料是否齊全、整體的密封性以及無菌連接器輻照滅菌驗證資料是否齊備。因為過濾的時候要考量濾器前后端的壓差,所以也要有相應的壓力監測系統,還要有相應的記錄及進行相應的計量。
(五)一次性系統。根據EU GMP規范,使用前要進行完整性測試。這時候要考量的是在無菌狀態下,做完整性測試過程中的通氣、通水經過相應的過濾,應當怎樣收集過濾的水。
(六)微生物負荷。需要重點考察的是取樣量。法規要求是10CFU / 100ml。ADC和其他大分子藥物附加值比較高,通常會采用折中的方案。例如取3ml,規定定量的CFU。微生物負荷的取樣時間,也是一個考量點。一般的取樣會在除菌過濾前。如果除菌過濾是持續的過程,那就要在除菌過濾前的最后時間點進行取樣。
四、生物凈化
第四部分就是ADC制劑生產過程中微生物控制的非常重要的一部分,即生物凈化,通常指隔離器VHP。
隔離器VHP需要重點關注四個方面:
(一)CD/CV的開發。隔離器的溫度、濕度、壓差以及蒸發盤的溫度、過氧化氫的注入量、持續時間,都是在CD/CV開發過程中重點考量的參數。
(二)BI的選擇與處理。BI的選擇重點考慮D值;BI的保存要注意溫濕度以及避免裸手接觸BI。驗證過程可考慮分數法,每個點布置3個BI,防止出現個別陽性的情況。
(三)VHP實施過程中的控制。要關注幾個點:一是VHP的起始階段,即除濕后的濕度。還要關注VHP過程中(提升階段)過氧化氫達到的濃度,該濃度需達到一定的數值,且經過驗證。如驗證的時候達到400PPM才能達到殺滅的效果,但如果實際過程中,濃度沒有達到400PPM則會造成風險,這就不是成功的VHP過程。凈化階段持續時間也很重要,比如驗證過程中進行的是85分鐘的驗證,生產過程中就會更保守,延長時間,真正對無菌保證起到雙保險的作用。
(四)過氧化氫排殘。一般要配備高低濃度探頭,有的設備商只配了一種探頭,并宣稱高低濃度均可監測。實際上這種探頭對高濃度比較精確,而低濃度時誤差非常大,所以并不適宜。比較適宜的做法是分開設置濃度探頭,并且要確認低濃度探頭的精度范圍。很多時候,低濃度探頭在1ppm的精度不高,排殘的時候顯示到了1ppm,如果以更精確的便攜式監測的話,可能有幾個ppm,這對于敏感性高的藥品有一定影響。同時,低濃度探頭的安裝位置也很重要。
在排殘時需要在驗證過程中做實驗,保證過氧化氫的濃度達到實際的數值,而不是在隔離器本身的探頭里面顯示的數值。因為探頭精度有限,一些特殊藥品對過氧化氫的敏感度非常高,此時需要單獨考察,考察時要做到:一是延長排殘時間,二是以相應的方法進行確認。