您當前的位置:檢測資訊 > 法規標準
嘉峪檢測網 2022-11-26 14:19
元素雜質又稱重金屬,重金屬原義指比重大于5的金屬,元素雜質包括可能存在于原料、輔料或制劑中,來源于合成中催化劑殘留、藥品生產制備過程中引入或輔料中存在的、生產設備引入、或容器密閉系統引入。某些元素雜質不僅對藥品的穩定性、保質期產生不利影響,還可能因為潛在的毒性引發藥物副反應。
因此歐盟、美國對雜質的控制越來越嚴格,對此項不斷修訂,中國在加入ICH后對此項檢測應該也會向國際靠攏,因此了解法規對元素雜質的要求、建立有效的檢測方法變得尤為重要。
一 USP對元素雜質的修訂歷程
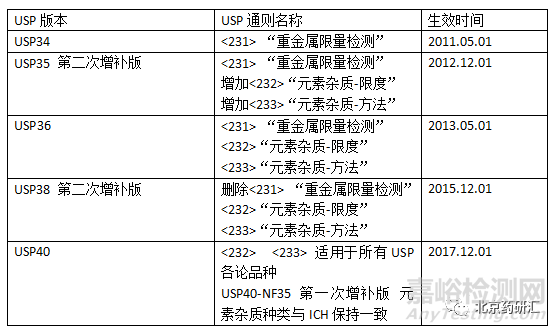
FDA規定在2018年1月1日之后,針對USP藥典品種,提交新的NDA、ANDA應該符合USP<232>、<233>。針對非USP藥典品種,申請人提交新的NDA、ANDA時,應該遵循Q3D。美國對元素雜質的規定與ICH規定在不同時期,內容不一致,但從2017年12月之后,USP對元素種類和限量均與ICH保持一致。修訂歷程詳見下表。
二 哪些產品需要控制?
本指南適用于化藥制劑產品以及生物藥產品,包括含有純化后的蛋白質和多肽(包括采用復合或非復合來源生產的蛋白質和多肽)的藥品及其衍生物、合成多肽、多核苷酸和低聚糖的藥品。
不適用的藥品包括不適用于臨床研究階段的藥品,也不適用于植物藥、放射藥、疫苗細胞、代謝產品、DNA產品、血制品、滲析溶液、產品中元素有療效的藥品,還有用于基因治療、細胞治療、組織再生的藥品以及保健品、獸藥、全靜脈營養液。
三 元素雜質內容簡介
因為目前美國和歐洲的指導原則,美國和歐洲的藥典對元素雜質的要求,基本和ICH保持一致。ICH對元素雜質要求有很多專家做過詳細的分析,所以本文以ICH對元素雜質要求做簡單的分析。
ICH對元素雜質的分類,如下:
Q3D元素雜質指南為口服、注射和吸入制劑中的24個元素雜質建立了允許日暴露量(Permitted Daily Exposure,簡稱PDE)
第1類
元素砷(As)、鎘(Cd)、汞(Hg)和鉛(Pb)是對人有毒性的物質,藥品生產中不得使用或限制使用,通常來源于礦物賦形劑。因此,所有給藥途徑的風險評估中都必須評價這4種元素。
第2類
本類別中的元素一般被認為是與藥物的給藥途徑有關的物質,又分A,B兩類。
2A類:元素鈷(Co)、鎳(Ni)、釩(V)。在藥品中出現可能性相對較高的元素,因而需要對所有元素雜質的潛在來源及所有攝入途徑(如所指)進行風險評估。
2B類:元素銀(Ag)、金(Au)、銥(Ir)、鋨(Os)、鈀(Pd)、鉑 (Pt)、銠(Rh)、釕(Ru)、硒(Se)、鉈(Tl),這些元素在自然界中稀少,在藥品中出現的可能性較低。除非其在原料藥、輔料或藥品的其它成分生產中被有意加入,否則可被排除在風險評估以外。
第3類
這類元素包括鋇(Ba)、鉻(Cr)、銅(Cu)、鋰(Li)、鉬(Mo)、銻(Sb)、錫(Sn)。口服途徑給藥,除非在原料藥、賦形劑或藥品的其他組分生產中有意添加,否則不需要在風險評估中進行考慮。對于生產中非有意添加的這些元素,鋰、銻和銅在注射和吸入給藥時需進行風險評估;鋇、鉻 、鉬和錫僅需在吸入給藥時進行評價。
其他元素:
由于固有毒性低和區域法規差異,沒有規定PDE,在該指南中未予以闡述。如果這些元素出現在藥品中,按照其他指南或區域法規進行處理。該類元素包括鋁(AL)、硼(B)、鈣(Ca)、鐵(Fe)、鉀(K)、鎂(Mg)、錳(Mn)、鈉(Na)、鎢(W)、鋅(Zn)。
因為美國藥典舊版出現過元素雜質種類、限度和ICH不一致的情況,如表4,很多相關分析文章對此進行了詳細的比較。 但是,目前新版USP的規定變更為和ICH一致,該情況應該引起注意。新版USP和ICH對規定元素種類和限度如表5(部分內容,具體規定參見ICH Q3D)。
三 無機元素雜質必須定入產品質量標準嗎?
PDE值的30%用來衡量是否將無機元素雜質定入藥品的質量標準。如果藥品中所有來源的元素雜質水平低于PDE值的30%水平,而申報人已對數據進行了適當的評估,證明對元素雜質的控制已經足夠充分,則不需要制定質量標準。
如果風險評估證明元素雜質的水平高于30%PDE,則要建立質量標準來保證元素雜質水平不會超過藥品的PDE值。
在提交申報時,如果沒有其它論證,一種元素雜質的水平和可變性可以通過提供成分或藥品具代表性的、3批生產規模或6批中試規模批次所得的數據來建立。對于有些具有內在可變性的成分(例如,礦物質輔料),在應用控制閾值時,除了30%的PDE值,可能還需要提供額外的數據。
四 藥品的元素雜質檢測法
采用USP<233>或自己驗證過的分析方法來檢測成品中無機元素雜質的含量,來評估成品中的各個無機元素是否滿足USP/ICH的PDE限度。
每日PDE≥測量值(ug/g)X每天最大劑量(g/day)
這個方法的主要優點是直觀,并考慮到了潛在的工藝設備產生的金屬污染。
五 元素雜質的計算方法
1 總量計算法
采用USP<233>或自己驗證過的分析方法來檢測各個原輔料組分中無機元素雜質的含量,按照每個組分的實際用量和藥品的最大日用量進行計算,來評估成品中的各個無機元素是否滿足USP/ICH的PDE限度。
其中:M=制劑中的每個組分
CM=該組分中重金屬濃度測量值(µg/g)
WM=制劑中組分的重量(g)
DD=每日給藥最大劑量(g/day)
2 單組分評估法
對藥品每日劑量不超過10g的品種,如果在制劑中每個原輔料組分的重金屬濃度不超過USP<232>中的表3或者ICH Q3D的表A.2.2中的濃度限度,說明成品中的各個無機元素滿足USP/ICH的PDE限度,不需要進一步計算。
方法1和方法2的優缺點如下:
主要優點:
(1)單組分樣品處理比制劑樣品處理更簡單;
(2)可以避免單個組分重金屬超標導致成品元素雜質超標;
(3)單一組分的測定結果可以用于多個制劑品種的計算。
主要缺點:
(1)需要對成品中每個組分進行檢測,分析工作量巨大;
(2)如果單一組分有多個供應商,需要對每個供應商提供的該組分進行檢測;
(3)沒有考慮到潛在的工藝設備產生的金屬污染。
六 結語
如何評估內包材中的無機元素雜質對液體和半固體制劑的影響?從內包材中可浸出的(leached)元素雜質:對可能從內包材引入的潛在元素雜質應基于藥品類型和其它包裝材料間的可能的相互作用進行評估。如果通過對結構材料的審核證明內包材不含有任何元素雜質,則不需要進行額外的風險評估。
由于無機元素從內包材浸出至固體劑型的可能性是非常小的,不需要在風險評估中進行深入考慮。但對于液體和半固體劑型,在藥品的貨架期內,元素雜質從內包材中浸出到藥品中的可能性比較大,此時應對內包材中可能浸出(leached)的元素雜質進行研究(在清潔、滅菌、輻射后等),一般要在藥品的內包材評估中對該類元素雜質做重點論述。
總之,無論是FDA、USP還是ICH都已經明確的要求從2018年1月1日起對所有上市的、正在申報的、將要申報的藥品進行無機元素雜質的風險評估,來保證藥品的安全性。

來源:Internet


