您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2022-05-09 07:33
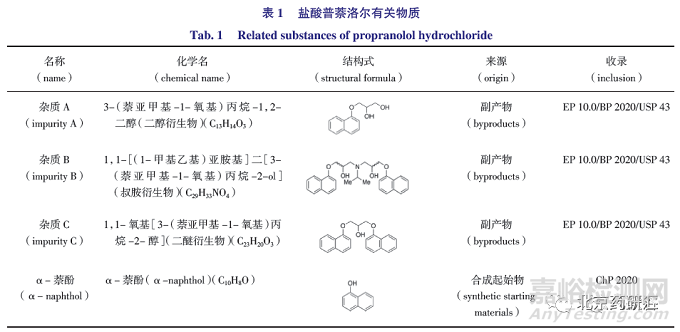
鹽酸普萘洛爾原料藥和片劑質量標準收載于《中國藥典》2020年版(ChP 2020),英國藥典2020/歐洲藥典10.0(BP 2020/EP 10.0)、美國藥典43(USP 43)和日本藥典(JP17)也均有收載。其中ChP 2020、BP 2020/EP 10.0、USP 43 和JP17 均采用液相色譜法檢查鹽酸普萘洛爾原料藥有關物質,色譜條件相同。ChP 2020 標準中有關物質采用主成分自身對照法,未對已知雜質進行單獨控制,JP 17標準與ChP 2020 限度控制基本相同,而BP 2020/EP 10.0 采用主成分自身對照法,USP 43 采用加校正因子的主成分外標法,對已知雜質A、B、C 進行單獨控制。各國藥典只有BP 2020/EP 10.0 收載的鹽酸普萘洛爾片標準中設定了有關物質檢查項,測定方法與原料藥相同。
本文在上述標準基礎上,對有關物質檢查方法進行了改進,改變了色譜條件,采用新型堿性色譜柱系統替代混合離子對方法,優化了有關物質檢測波長,篩選了合適的色譜柱并進行方法學驗證。同時,對在部分樣品中發現的較大的未知雜質進行了分析確證,并綜合結構和合成工藝分析該物質的可能來源。
1 鹽酸普萘洛爾的有關物質分析
經過對國內企業鹽酸普萘洛爾原料藥化學反應過程及工藝流程的調研發現,國內企業起始物料、合成路線基本一致,起始物料均為 α- 萘酚(甲萘酚),即現行標準中檢查的游離萘酚,合成主要步驟為醚化、胺化和成鹽,因而雜質差異不大,所用輔料均為常見的片劑輔料,因此在合成過程中可能存在副產物的產生。此外,鹽酸普萘洛爾片生產企業的處方略有不同,但加工工藝基本一致,采用常用的制粒壓片法。
BP 2020/EP 10.0和USP 43收錄的已知有關物質主要包括雜質A、雜質B 和雜質C,均來源于合成副產物,ChP 2020 檢查的游離萘酚,為合成本品的起始物,見表1。
1.1 現行標準方法檢驗
ChP 2020、BP 2020/EP 10.0、USP 43 和JP17 采用液相色譜法檢查本品有關物質,色譜條件相同。分別稱取鹽酸普萘洛爾對照品(中檢院)、雜質A、B、C 和α- 萘酚對照品,用乙腈- 水(50∶50)溶解并稀釋制成約含主成分1 mg·mL -1 ,已知雜質均約為1 μg·mL -1 的混合對照品溶液,作為系統適用性溶液(SST)。參照ChP2020 有關物質檢測方法,選擇Agela Technologies C18(250 mm×4.6 mm,5 μm)色譜柱,以乙腈- 水- 硫酸(55∶45∶0.1)的混合液(取十二烷基硫酸鈉 1.6 g 和磷酸二氫四丁基銨 0.31 g溶于1 000 mL 混合液中,用2 mol·L -1 氫氧化鈉溶液調節pH 3.3)為流動相,檢測波長為292 nm,進樣20 μL。結果如圖1 所示,混合對照品溶液中各組分峰形和柱效均不理想,α- 萘酚峰與普萘洛爾峰幾乎重疊。

由于α- 萘酚峰是鹽酸普萘洛爾在合成中使用的原料,嘗試調整有關物質色譜條件,將主成分和各已知雜質在同一色譜系統中實現分離。通過調整十二烷基硫酸鈉與磷酸二氫四丁基銨的濃度,以及換用庚烷硫酸鈉,均未能達到理想的分離效果,故對有關物質測定色譜條件進行了重新研究,建立了不使用離子對試劑的堿性流動相色譜系統。
1.2 新建方法的色譜條件確定
1.2.1 流動相pH
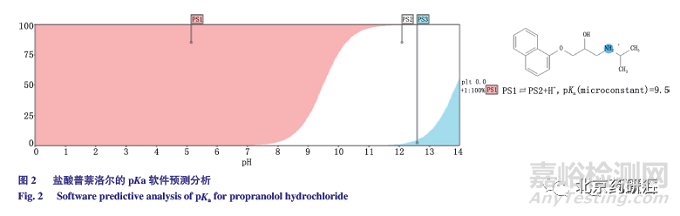
經軟件ChemDraw 預測分析,鹽酸普萘洛爾的pKa 為9.5(圖2),為兼顧主峰的峰形以及色譜柱的耐受性,采用醋酸銨為緩沖鹽,配制10mmol·L -1 醋酸銨溶液,調節流動相中醋酸銨溶液的pH 為10.0、10.5、11.0 和11.3。為進一步提高雜質分離度,改為梯度洗脫。
由表2 可知,流動相中水相的pH 對色譜峰的拖尾因子有影響,pH 越低,拖尾因子越大。在該色譜條件下,主峰前發現一未知雜質,與主峰相鄰,pH 10和10.5 時的主峰與相鄰雜質不能達到基線分離,pH11.3 時的分離度最優,然而pH 過高則對色譜柱的耐堿性要求更高,綜合評價pH 11.0 最優。

1.2.2 流動相比例
為進一步提高雜質分離度,色譜條件其他參數不變,僅改變梯度洗脫程序初始流動相的水相和有機相比例,分別設為醋酸銨溶液(10 mmol·L -1 醋酸銨溶液,用濃氨溶液調節pH 至11.0)- 乙腈(90∶10)-乙腈(67∶33)、(70∶30)和(73∶27)。由表3 可知,當比例為73∶27 時,主峰與相鄰峰分離度和拖尾因子最好,然而流動相中水相比例增加,主峰保留時間延長,處于梯度變化時間內,有可能影響未知雜質的檢出,綜合考慮流動相比例確定為70∶30。
1.2.3 色譜柱
優化后色譜條件使4 種已知雜質和主成分均能有效分離,不同品牌色譜柱對色譜峰的分離度影響比較大,篩選具有高效分離而且耐堿性的色譜柱可能會提高分離度。分別選擇CERIL-Column3、TOSOH TSK gel ODS-80TS 和WatersXBridgeTM 的C18 色譜柱,由表4 可知,選擇的3 種不同品牌相同規格的色譜柱除與主峰相鄰的未知雜質之外,其他雜質均能很好分離。TSK gel ODS-80TS 和XBridgeTM 色譜柱,均不能將主峰與該相鄰雜質分離,而TSK gel 色譜柱也延長了主峰的保留時間。經過篩選,CERI 的L-column3 C18 色譜柱能很好地分離相鄰雜質和主峰。因此本方法只能選擇分離度高的耐堿性色譜柱,推薦CERI L-Column3 色譜柱或效能相當的色譜柱。

1.2.4 檢測波長
各國藥典標準檢測波長均選擇292 nm,在上述流動相條件下采用PDA 檢測器對SST 中的鹽酸普萘洛爾、雜質A、雜質B(顯示2 個相連的色譜峰,為同分異構體)、雜質C、α- 萘酚及與主成分的色譜峰相鄰未知雜質在190~400 nm 范圍內進行光譜掃描。由表5 可知,上述化合物的最大吸收峰集中在213~230 nm 的范圍內,292 nm 波長處的吸收均較弱,目標化合物在213~230 nm 的范圍內的吸收度遠大于292 nm 處,綜合考慮梯度洗脫對基線的影響等因素,最終選擇230 nm 作為本方法的檢測波長。

1.3 色譜條件的確定
根據上述對色譜條件的探索,確定鹽酸普萘洛爾片的有關物質的檢測方法如下:色譜柱:CERI L-column3 C18(250 mm×4.6 mm,5 μm);流動相A:醋酸銨溶液(10 mmol·L-1 醋酸銨溶液,用濃氨溶液調節pH 至11.0)- 乙腈(90∶10);流動相B:乙腈;檢測波長:230 nm;柱溫:30 ℃;流速:1.2 mL·min -1 ;進樣體積:20 μL;梯度洗脫程序見表6。

取“ 2.3”項下方法制備ST 溶液,照色譜條件試驗,在230 nm 波長進行檢測,各已知雜質和主峰相鄰未知雜質均可以達到有效分離,見圖3。

1.4 與主成分色譜峰相鄰未知雜質的結構研究與毒性預測
1.4.1 結構研究
由于所建立的HPLC 檢測方法采用了更靈敏的檢測波長,且具有更好的分離效率,使得在鹽酸普萘洛爾主峰前1 個新未知雜質被檢出,該雜質在各國藥典中均未提及,但在中檢院提供的鹽酸普萘洛爾對照品以及部分樣品中有檢出,且為最大單個雜質,如圖4 所示,該未知雜質的最大吸收波長為225 nm,在292 nm 處幾乎沒有吸收,因此使用現行標準中292 nm 檢測波長未能檢出該雜質。
采用上述液相色譜條件,通過質譜檢測器對未知雜質的結構進行了分析,其與鹽酸普萘洛爾的一級質譜和二級質譜見圖5。通過LC-MS 研究發現,該未知雜質不僅與鹽酸普萘洛爾有相近的保留時間,其一級質譜和二級質譜也非常接近,僅二級質譜的碎片離子豐度有一定差異,故推測其應為鹽酸普萘洛爾的同分異構體,且結構相似。

結合紫外光譜信息和鹽酸普萘洛爾的合成工藝初步推測未知雜志結構為側鏈接在萘環β 位所形成異構體見圖6,該雜質來自合成原料α- 萘酚中混入的β- 萘酚的反應產物。
依據上述推斷,進一步以β- 萘酚為底物合成了β- 同分異構體,其在LC-MS 上的保留時間和一級質譜、二級質譜信息上與未知有關物質完全一致。進一步通過質譜、核磁共振等方法對所合成的β-同分異構體進行了結構確證,所有結果與推測完全一致見圖7。依據鹽酸普萘洛爾的結構和合成工藝分析,正常的合成和貯存過程產生β- 位的異構體可能性微乎其微,合成底物α- 萘酚(鹽酸普萘洛爾的合成原料)中混有的β- 萘酚應為產生β- 異構體主要來源。

1.4.2 毒性預測
查閱相關資料,目前尚缺乏對未知雜質β- 同分異構體藥理毒理研究資料,因此本研究使用Simulations Plus 公司的ADMET Predictor計算軟件對該雜質進行了毒性預測。預測結果顯示該雜質口服急性毒性低,表現出一定的對梨形四膜蟲毒性,魚類毒性、蜜蜂毒性,無染色體變異性,不會產生致突變性,不會致癌,大鼠急性毒性低,無其他明顯毒性,在體內不可降解。預測結果表明,該雜質的毒性均較小,但仍需進一步進行藥理毒理性質研究,該雜質的存在具有一定的潛在風險,應予以控制。
2.6 方法學驗證
2.6.1 耐用性試驗
取上述SST 溶液,色譜條件其他參數不變,分別考察該方法在不同柱溫(27、30、35 ℃)、不同流速(1.1、1.2、1.3 mL·min -1 )、不同批次CERI L-Column 3(250 mm×4.6 mm,5 μm)色譜柱的耐用性。
不同柱溫的影響:由表7 可知,溫度在27 ~35 ℃范圍內變化,色譜峰特性略有變化。溫度升高保留時間遷移,主峰與相鄰雜質峰分離度和理論板數也有所升高,雜質B 相鄰雙峰分離度略有下降,拖尾因子略有降低,綜合評判柱溫設置為30 ℃,主峰與相鄰雜質峰分離度滿足基線分離。

不同流速的影響:由表8可知,流速在1.1 ~1.3 mL·min - 1 范圍內變化,色譜峰特性略有改變,隨著流速增加,主峰保留時間縮短,主峰與相鄰雜質峰分離度和雜質B 相鄰雙峰分離度略降低,拖尾因子也隨著降低,綜合評判采用1.2 mL·min -1 流速。
不同批次色譜柱的影響:由表9 可知,研究采用的CERI 品牌不同批號色譜柱影響不大,均可以將β- 同分異構體與主峰基線分離。

2.6.2 專屬性試驗

2.6.3 重復性試驗
(1)如表9 所示,6 份供試品均檢測出雜質B,結果在0.026%~0.029%,其它已知雜質均未檢出;6 份樣品中其他未知最大雜質含量在0.011%~0.018%,雜質總量均小于0.1%,各雜質含量基本一致,重復性良好,符合有關物質精密度試驗要求。

(2)如表11 所示,6 份供試品均檢測出雜質B,結果在0.028%~0.031%;均在主峰前發現β- 異構體峰,含量在0.326%~0.359%,其他已知雜質A、B、α- 萘酚均未檢出;6 份樣品其他單個最大雜質均在0.012%~0.018%。因為該樣品中檢出β- 異構體,雜質總量在0.393%~0.427%,各雜質含量基本一致,重復性良好,符合有關物質精密度試驗要求。

2.6.4 溶液穩定性試驗
取上述重復性試驗供試品溶液,分別在下述時間點進樣分析,記錄色譜圖,考察雜質峰面積變化情況。
(1)如表12 所示,0~48 h 內不同時間點進樣鹽酸普萘洛爾原料藥供試品溶液均檢測出雜質B,含量結果在0.027%~0.029%,其他已知雜質均未檢出;其他單個最大雜質含量在0.010%~0.028%,無新的雜質檢出,雜質峰面積之和雖略有變化,但是顯示和放置時間關系不大,雜質總量未超過0.1%。結果表明,供試品溶液在48 h 內可保持基本穩定。


(2)如表12 所示,不同進樣時間鹽酸普萘洛爾片供試品均檢測出雜質B,結果在0.028%~0.031%;均在主峰前發現β- 異構體峰,含量在0.329%~0.337%,其他已知雜質A、C 及α- 萘酚均未檢出,供試品溶液在48 h 內可保持穩定,沒有新的雜質檢出,符合有關物質試驗要求。
2.6.5 定量下限和檢測下限
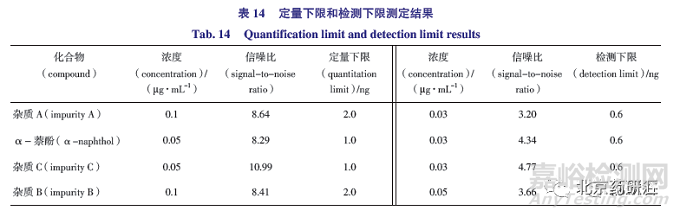
取鹽酸普萘洛爾對照品(USP)和已知雜質對照品各適量,分別用乙腈-水(50∶50)溶解并定量稀釋,精密量取20 μL 進樣,注入色譜儀,記錄色譜圖。當信噪比約為10∶1 時測得鹽酸普萘洛爾定量限為4 ng(0.2 μg·mL -1 ),相對供試品濃度為0.02%;當信噪比約為3∶1 時測得檢測限為2 ng(0.1 μg·mL -1 ),相對供試品濃度為0.01%,檢測限濃度約為對照品溶液濃度(1 μg·mL -1 )的1/10,靈敏度可滿足測定需求,其他已知雜質的定量限和檢測限結果見表13。
2.7 樣品檢測
2.7.1 鹽酸普萘洛爾原料藥
采用擬定方法檢測3 家企業提供的3 批樣品,結果見表15 和圖9,其中企業-2 和企業-3 的單個雜質和雜質總量均在0.1% 以下,而企業-1 由于含有β- 異構體,含量為0.347%,雜質總量為0.389%。

2.7.2 鹽酸普萘洛爾片
采用擬定方法檢測參比制劑和3 家仿制藥企業共4 批樣品,結果見表16 和圖10,其中參比制劑不含β- 異構體,雜質總量僅為0.048%,仿制藥企業4 不含β- 異構體,而仿制藥企業5 和6 均檢出β- 異構體,含量分別為0.023% 和0.346%,仿制藥企業4 和5 單個雜質和雜質總量均低于0.1% 和0.2%,而企業-6 由于含有β- 異構體,雜質總量為0.457%。


3 討論
3.1 新的有關物質檢測方法的建立
目前,已報道測定鹽酸普萘洛爾的分析方法有色譜法、毛細管電泳法、電化學分析方法、光纖傳感分析技術等。色譜法具有分離效果好、輔料無干擾 、快速、簡便等優勢,已廣泛應用在鹽酸普萘洛爾的檢測中。各國現行版藥典均采用復雜的混合離子對色譜法檢查鹽酸普萘洛爾有關物質,該方法存在的問題是,實測發現峰形和柱效均不理想,對普萘洛爾的結構類似物(如α- 萘酚、β- 酚異構體)不能有效分離,且292 nm 的檢測波長靈敏度十分有限。本探索研究建立了新的HPLC 測定方法,用新型堿性色譜柱系統替代混合離子對方法,并采用更為靈敏的230 nm 波長進行檢測。該方法不僅對歐洲藥典收載的3 種已知雜質(有關物質A、B、C)可以有效分離,同時對普萘洛爾的結構類似物具有更好的分析能力。并且本方法流動相水相的pH 為11.0,因此需要選擇耐堿性比較強的色譜柱。通過篩選比較,CERI 生產的L-column3 C18 色譜柱采用了獨特的氣態峰尾技術和新開發的PCS 硅膠技術,穩定性較好,適用的pH范圍為1~12,具有較高的耐堿性。研究也證實該色譜柱的不同批次均表現較好的穩定性和耐受性,因此推薦操作中采用該色譜柱或效能相當的色譜柱。
經對不同生產企業原料藥和片劑樣品檢測以及同批次樣品在不同的保存條件下的樣品檢測結果進行分析,各特定雜質、單個最大雜質和雜質總量均無顯著性差異,進一步表明本品有關物質主要來自原料藥,與制劑生產工藝和保存條件關系不大,風險控制的重點應控制原料藥。
3.2 β- 異構體研究
根據新建的有關物質檢測方法,發現原料藥生產企業-1 含有β- 異構體,而2 家仿制藥生產企業5 和6 也檢出此雜質。經調研發現,此2 家仿制藥生產企業的原料藥來源均為原料藥生產企業-1,其中仿制藥生產企業-6 和原料藥生產企業-1 為同1 批次,2 者β- 異構體含量相似,表明該雜質在制劑加工中未有變化,而仿制藥生產企業-5 原料藥來源為不同批次,β- 異構體含量較低,表明該原料藥生產企業合成工藝不穩定,導致各批原料間β- 異構體含量不一致。依據鹽酸普萘洛爾的結構和合成工藝分析,正常的合成和貯存過程產生β- 異構體可能性微乎其微,在強制破壞性試驗中也未產生β- 異構體,因此推測合成底物α- 萘酚(鹽酸普萘洛爾的合成原料)中混有的β- 萘酚應為產生β- 異構體主要來源。由于目前尚缺乏對β- 異構體藥理毒理研究資料,故應對β- 構異構體的潛在風險予以關注,并設法控制其限量,進一步研究β- 異構體性質,制定更為科學的限度。根據新建的檢測方法,發現參比制劑檢測并無此雜質,而國內中檢院提供對照品以及部分樣品均含有此雜質,應在對照品制備和仿制藥評價中予以關注。此外,考慮到β- 萘酚的反應位阻較低,類似的情況在其他使用α 酚萘酚作為合成底物的藥物中也可能存在,應同樣加以重視,同時對作為原料藥合成底物的α- 萘酚應制定更為嚴格標準,從源頭上控制產生此類雜質的潛在風險。
4 結論
本研究對鹽酸普萘洛爾原料藥和片劑現行標準中的有關物質測定方法進行了考察及優化,在此基礎上建立的新方法靈敏度高,專屬性強,操作簡便,能將游離萘酚檢查納入有關物質檢測中,并有效檢出現行標準未能檢出的β- 異構體,為《中國藥典》2020 年版中鹽酸普萘洛爾質量標準的修訂提供了參考。綜合結構和合成工藝分析,推測該物質為鹽酸普萘洛爾的β- 異構體,是鹽酸普萘洛爾的合成底物α- 萘酚中混有β- 萘酚而產生的合成副產物,由于該β-異構體藥理毒理性質尚無相關詳細資料,該雜質的存在具有一定的潛在風險。通過研究,還確認了鹽酸普萘洛爾高風險雜質及質量控制關鍵點,指導企業優化相應的生產工藝,提升產品質量,為科學監管提供技術保障和執法依據,促進國產仿制藥質量的不斷提高,為一致性評價工作提供有力的技術支撐。

來源:Internet


