您當(dāng)前的位置:檢測資訊 > 科研開發(fā)
嘉峪檢測網(wǎng) 2022-01-19 22:00
導(dǎo)讀
科學(xué)的審評理念和監(jiān)管模式一直以來是美國FDA孜孜不倦追求的目標(biāo)。經(jīng)過百年的發(fā)展,F(xiàn)DA已經(jīng)形成一套先進(jìn)監(jiān)管制度。本文以Emflaza為例,重點關(guān)注技術(shù)審評對非臨床研究完整性及橋接臨床研究,介紹并分析FDA的關(guān)注點及審評思路,希望對今后新藥研發(fā)和申報提供借鑒信息。
FDA審評里程碑
2016年6月9日,MarathonPharmaceutical向FDA遞交了deflazacort(地夫可特)片劑和口服溶液上市申請數(shù)據(jù),劑量為0.9mg/kg/日,每日一次。片劑規(guī)格包括6,18,30,35mg,口服溶液規(guī)格為22.75mg/mL,13ml,20ml/瓶。申報的適應(yīng)癥為DMD。商品名為EMFLAZATM。
pre-NDA 會議于2015年8月4日召開,F(xiàn)DA認(rèn)為研究資料足以支持本品的上市申請。2016年6月9日首次正式書面提交505(b)2申請,PDUFA為2017年2月9日。
Emflaza在審批過程中獲得一系列優(yōu)惠待遇,被授予快速審查資格 (fasttrackdesignation),用優(yōu)先審批程序(priority review)進(jìn)行審批,此外Emflaza還被 FDA授予孤兒藥資格和“罕見兒科疾病治療藥物優(yōu)先審批”(Rare Pediatric Disease Priority Review Voucher)資格,這是FDA在2007年推出這一項目以來第9個獲此待遇的兒科藥物

產(chǎn)品背景信息
地夫可特的制劑沒有在美國獲得批準(zhǔn),但是在全球其他國家獲得批準(zhǔn)作為糖皮質(zhì)激素治療自身免疫性疾病和過敏反應(yīng),2008年在英國獲批,2013年在瑞士獲批,1990年在西班牙獲得批準(zhǔn),以及德國,希臘,意大利,葡萄牙,墨西哥,加勒比,印度,韓國等。Sanofi在全球多個國家上市的地夫可特商品名為Calcort®。地夫可特在美國沒有獲得任何適應(yīng)癥的批準(zhǔn)。雖然地夫可特和潑尼松沒有被正式批準(zhǔn)用于治療DMD,但是美國神經(jīng)病學(xué)會(AAN)和疾病控制中心(CDC)指導(dǎo)原則都推薦糖皮質(zhì)激素作為DMD患者改善肌肉強度和功能的一線治療,臨床也常超說明書使用來延緩肌肉強度喪失。
體外研究表明地夫可特在血漿乙酰水楊酸酯酶作用下快速轉(zhuǎn)化為活性形式21-desDF1,然后由CYP3A4作用轉(zhuǎn)化為其他代謝物質(zhì)。臨床研究表明6β-OH-DFZ是主要代謝物(代謝物質(zhì)III)約占總體的27% 。文獻(xiàn)資料報道代謝物V(分子量417)也是主要循環(huán)代謝物,占總體的25%。但是文獻(xiàn)報道中沒有對代謝物V的結(jié)構(gòu)組成尚不清楚,而且申請人沒有在后續(xù)的臨床藥理研究中對代謝物V進(jìn)行測定。
不充分的主要代謝物非臨床研究數(shù)據(jù)
支持本品的非臨床數(shù)據(jù)包括申請人開展的8項非臨床研究和文獻(xiàn)報道。
藥理學(xué)研究
申請人開展21-desDFZ和代謝物6β-OH-21-desDFZ的主要藥理學(xué)研究,以及CHO-K1細(xì)胞中進(jìn)行的hERG試驗以評估兩種代謝物21-desDFZ and 6β-OH-21-desDFZ的安全藥理研究。最高試驗濃度(10um)時,對hERG電流的抑制作分別達(dá)到~8%和~14%。因此沒有發(fā)現(xiàn)兩種化合物有QTc 延長的信號。
PK/ADME
地夫可特和21-desDFZ的PK/ADME體內(nèi)研究,以及體外研究評估21-desDFZ血漿蛋白結(jié)合和代謝的穩(wěn)定性。體外蛋白結(jié)合試驗結(jié)果21-desDFZ與人血清蛋白和α-酸性糖蛋白結(jié)合率低(分別為~55和20%)。兩項體外試驗研究21-desDFZ的代謝途徑,21-desDFZ與大鼠,狗,人的肝微粒體培養(yǎng)。人肝微粒體中代謝程度最高,大約~83,85,60分鐘培養(yǎng)結(jié)束時剩余40%21-desDFZ。人肝微粒體形成7種代謝產(chǎn)物。
毒理學(xué)研究
申請人沒有進(jìn)行地夫可特的急性毒性研究,急性毒性引用了文獻(xiàn)數(shù)據(jù)。致死劑量>4000mg/kg。申請人開展的地夫可特和代謝物21-desDFZ體外和體內(nèi)(大鼠和猴子)的重復(fù)毒性研究。Sprague-Dawley大鼠胃管灌食法分別給予0.05,0.15,或0.50mg/kg/日,每日一次。雄性劑量≥0.05 mg/kg/日組,雌性劑量≥0.10mg/kg/日組出現(xiàn)皮膚癥狀,行為改變,體重降低的反應(yīng)。雄性劑量≥0.05 mg/kg/日組,雌性劑量1.00 mg/kg/日組觀察到食量下降。劑量摸索基于14天劑量摸索研究(1/sex/group),給藥劑量為0,0.3,1.0,3.0mg/kg;NOAEL為3.0mg/kg
生殖和發(fā)育毒性
DMD患者幾乎全部是男性,因此無需進(jìn)行標(biāo)準(zhǔn)生殖和發(fā)育毒性研究。8周幼崽動物的毒理研究評估出生后發(fā)育過程中地夫可特的不良作用,以支持小于12歲的DMD患者人群。生殖毒性研究表明大鼠和猴子給藥地夫可特后,雄性動物的生殖力和發(fā)育的終點指標(biāo),沒有與地夫可特相關(guān)的睪丸損傷或精子評價指標(biāo)的變化。
遺傳毒性
申請人研究地夫可特和其代謝物的遺傳毒理學(xué)研究,包括地夫可特的標(biāo)準(zhǔn)遺傳毒理學(xué)研究(Ames,人淋巴細(xì)胞染色體畸變體外研究,大鼠體內(nèi)微核試驗);代謝物21-desDFZ的體外遺傳毒理試驗(Ames,人淋巴細(xì)胞染色體畸變)。
致癌性
pre IND會議申請人與FDA達(dá)成一致,在4期研究中開展致癌性研究,因此NDA申請中研究者沒有進(jìn)行地夫可特的致癌性
FDA對于非臨床數(shù)據(jù)的審評意見
FDA認(rèn)為已有非臨床數(shù)據(jù)和文獻(xiàn)資料缺少主要代謝產(chǎn)物:
6β-OH-21-desDFZ和 M-V的遺傳毒性和致癌性研究數(shù)據(jù),M-V的長期毒性數(shù)據(jù)。申請人需要補充開展體外遺傳毒性試驗(Ames,體外染色體畸變)和體內(nèi)(微核)試驗,6β-OH-21 – desacetyl - deflazacort和 M-V的致癌性研究。M-V的長期毒性研究。考慮到本品適應(yīng)癥的特殊性,F(xiàn)DA認(rèn)為可以作為上市后研究開展。
臨床三期試驗的橋接研究
90年代初期開展的2項三期臨床研究(MP-104-NM-001,MP-104-NM-002)支持本品有效和安全性。MP-104-NM-001是一項隨機(jī)雙盲,安慰劑和活性藥物對照研究,評估地夫可特在5-15歲DMD男性患者中改善肌力的療效(n=196)。受試者隨機(jī)接受地夫可特0.9/mg/kg/天,或1.2/mg/kg/天,潑尼松0.75 mg/kg/天,和安慰劑。地夫可特兩個劑量組均較安慰劑組有顯著改善。MP-104-NM-002是一項支持性研究評價地夫可特在5-11歲DMD男性患者的療效(n=38)。受試者服用地夫可特2mg/kg,隔日給藥,或者安慰劑。與安慰劑組相比地夫可特組在6個月時肌力提高(LS平均改變0.57),安慰劑組(LS平均改變-0.64,p=0.019)。
由于兩項臨床研究均在90年代初期進(jìn)行,臨床藥品的處方信息無法獲得。由于3期臨床研究沒有收集PK數(shù)據(jù)。因此無法直接橋接3期臨床研究數(shù)據(jù)。因此申請人進(jìn)行了一系列BA/BE試驗希望間接橋接3期臨床研究。
申請人進(jìn)行BA/BE研究包括:地夫可特片6mg多劑量研究;地夫可特片6*6mg單劑量;禁食,高脂肪餐后,研碎后與蘋果汁混合分別服用地夫可特片36mg,禁食服用地夫可特片6*6mg;禁食服用地夫可特混懸液36mg的生物等效性和食物影響研究;擬申報EMFLAZA (36mg)與Calcort (6*6mg)的生物等效性研究。為了橋接早期進(jìn)行的臨床研究,避免重復(fù)研究,申請人進(jìn)行了多項BA/BE試驗,最終獲得FDA的批準(zhǔn)。我們分析一下申請人如何解讀這些數(shù)據(jù)來支持橋接3期研究的。

首先各個處方的臨床藥理研究顯示不同處方結(jié)果都符合生物等效性限度要求,因此可以認(rèn)為地夫可特吸收對處方差異不敏感。其次食物影響研究中高脂肪餐沒有影響地夫可特的暴露程度(AUC)。因此可以推斷出pH和腸道內(nèi)容物不影響地夫可特在體內(nèi)的溶解和后續(xù)吸收。并且根據(jù)地夫可特物理化學(xué)性質(zhì),預(yù)估經(jīng)腸道吸收率很高(高于劑量的95%)。EMFLAZA與Calcort的BE研究證明二者生物等效。雖然美國沒有批準(zhǔn)Calcort,但是Calcort在多個國家都批準(zhǔn)了非DMD適應(yīng)癥。
3期臨床研究中不同給藥方案均觀察到療效。MP-104-NM-001地夫可特劑量方案包括0.9 mg/kg/日,1.2 mg/kg/日,每日一次。MP-104-NM-002地夫可特劑量方案包括2mg/kg,隔日給藥。這些劑量方案均觀察到對DMD患者的療效。地夫可特的劑量效應(yīng)曲線相對比較平坦(0.9,1.2mg/kg/日,2mg/kg/q2D),處方不同造成的微小的劑量差異不會影響臨床有效性。
綜合以上證據(jù),地夫可特獨特的性質(zhì)能夠保證藥物在體內(nèi)的吸收程度不受ph和處方而改變。申報處方的生物利用度不會低于臨床3期臨床研究處方。基于這些研究數(shù)據(jù)FDA認(rèn)為可以支持橋接3期臨床研究結(jié)果。
專利狀態(tài)
本次申請的地夫可特片和口服液兩種劑型沒有查詢到在美國有專利保護(hù),原研廠家在中國沒有相關(guān)專利申請。查詢到國內(nèi)企業(yè)申報的化合物及制備工藝的專利,9家企業(yè)申報地夫可特生產(chǎn)工藝,制備方法的專利,1家企業(yè)申報微粉化地夫可特口服制劑的專利。
國內(nèi)上市及申報情況
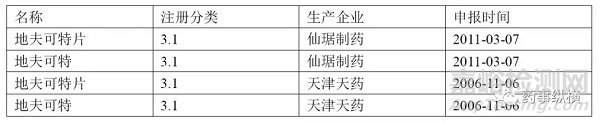
地夫可特原料及制劑目前在國內(nèi)沒有已經(jīng)批準(zhǔn)上市的產(chǎn)品。但是仙琚制藥和天津天藥2家公司已經(jīng)向藥品審評中心遞交了口服片劑的臨床研究的申請,同時關(guān)聯(lián)申報地夫可特原料藥。
杜氏肌營養(yǎng)不良及治療現(xiàn)狀
杜氏肌營養(yǎng)不良是一種X染色體隱性遺傳疾病,其特征是進(jìn)行性肌肉退化和萎縮,是肌營養(yǎng)不良中最常見的類型。DMD由抗肌萎縮蛋白(dystrophin)缺乏所導(dǎo)致,這種蛋白有助于保持肌肉細(xì)胞的完整性。DMD的發(fā)病年齡通常在3~5歲之間,并且會持續(xù)惡化,主要發(fā)生于男孩。據(jù)統(tǒng)計,全球平均每3500個新生男嬰中就有一人罹患此病。患者在學(xué)齡前就會因骨骼肌不斷退化出現(xiàn)肌肉無力或萎縮,導(dǎo)致不便行走。隨著病情的發(fā)展,將會出現(xiàn)可危及生命的心臟和呼吸系統(tǒng)疾病 ,大概在7歲到12歲時,會徹底喪失行走能力,患者通常在 20多歲或者30多歲時死于這些疾病,但疾病嚴(yán)重程度和預(yù)期壽命因個體而異。
除了本品Emflaza以外,eteplirsen(Exondys51)是FDA目前唯一批準(zhǔn)的治療DMD藥物,2016年獲得加速批準(zhǔn)用于治療編碼抗肌萎縮蛋白基因(dystrophin)突變中,針對略過第51個外顯子治療的一部分DMD患者。Eteplirsen獲得加速批準(zhǔn)是基于某些接受治療的患者骨骼肌中觀察到了抗肌萎縮蛋白的增加,達(dá)到了替代終點。Eteplirsen是首個經(jīng) FDA批準(zhǔn)用于治療該疾病的新藥,適用于確診抗肌萎縮蛋白基因外顯子 5l(exon51)突變的患者,這類人群約占DMD患者總數(shù)的 13%。此外糖皮質(zhì)激素,例如潑尼松,臨床也常超說明書使用來延緩肌肉強度喪失。

來源:Internet


