您當前的位置:檢測資訊 > 實驗管理
嘉峪檢測網 2022-01-18 20:27
大家在做試驗的時候,如果仔細查看色譜圖,就會發現幾乎每一個色譜峰都有一定程度的拖尾現象。有的比較嚴重,對有關物質檢查會造成一定的影響。
這本不是問題,但是當拖尾嚴重到一定程度,就會影響我們的分析結果,所以今天就讓我們來談一談關于液相色譜峰的拖尾問題。同時對氣相色譜峰拖尾產生的原因及解決方法,進行簡單的闡述。
什么是拖尾峰?
前沿陡峭,后沿較前沿平緩的不對稱峰,稱為拖尾峰。下圖就是一個拖尾峰,我們會發現這個峰的右半邊峰寬是大于左半邊峰寬的,尤其在基線處更加明顯。

那如何評價色譜峰的拖尾呢?絕大多數情況下制藥行業的從業人員會選擇用美國藥典(USP)中拖尾因子(Tf)來評價而其余的分析行業內人員則會選擇用對稱因子(As)來評價,拖尾因子和對稱因子到底有什么關系?
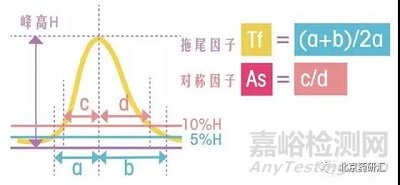
其中,拖尾因子Tf=(a+b)/2a,其中a和b分別是5%峰高處的左右半峰寬
對稱因子As=c/d,其中c和d分別是10%峰高處的左右半峰寬
如果a=b,c=d,那么拖尾因子和對稱因子的數值都等于1,也就是該峰不拖尾。實際工作中,當拖尾因子和對稱因子小于2時,這兩個值是非常接近的,如下圖所示。

拖尾峰會對工作造成什么影響?
1. 影響峰高的計算
先來看看第一個峰(Tf=1.0)和最后一個峰(Tf=3.5),它們的峰面積雖然是相同的,但是由于最后一個峰拖尾很嚴重,所以它們的峰高相差三倍。當我們使用峰高來計算最低檢出限時拖尾峰的負面影響就會比較明顯。
2. 影響峰面積的計算
當我們對色譜峰進行積分來計算峰面積時,峰的起點和終點通常情況下通過色譜峰的斜率來確定。色譜峰越拖尾,它的尾部基線越平緩,終點就越難確認。這一點在基線波動較大時體現的更加突出,那么它就會影響我們積分得到的峰面積。
3. 影響對某些痕量組分的確認
在藥典計算有關物質的相關要求中會提到,峰面積達到主峰面積0.1%的組分都要參與計算。
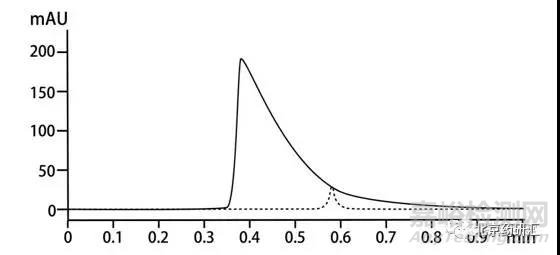
那么當一些小峰和前面的峰分離度不是很好時就會容易被隱藏在前面峰的拖尾處從而無法參與計算。
綜上所述,拖尾峰會對色譜分析的定性定量結果都會產生影響。
產生拖尾峰的原因是什么?
1、拖尾峰的形成是因為在色譜分離時出現了一種以上的保留機制,并且其中一種保留機制是超負荷的。在日常分析中大多數情況下會使用反相色譜。以常用的C18柱為例,C18是被鍵合在載體硅膠表面的,而硅膠是由硅和氧聚合而成的,所以它的表面會出現各種形態的硅羥基。
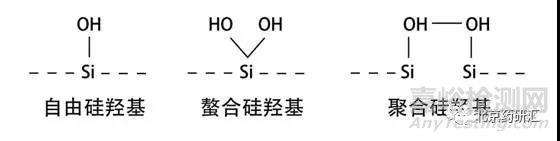
雖然理想的狀態是流動相中的樣品只和固定相C18發生作用。而實際上一半以上的硅膠表面并沒有被鍵合上C18,所以硅膠表面會裸露出大量的硅羥基(Si-OH)。
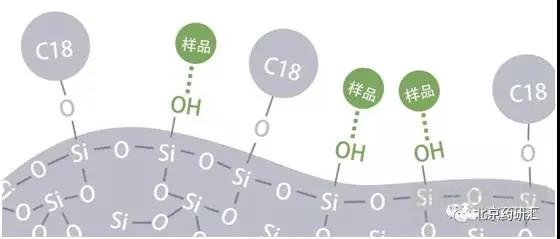
而單獨的自由硅羥基會和流動相中的樣品發生作用,形成拖尾。于是在最近的十幾年中,色譜柱供應商致力于生產出高純度的硅膠載體。由于新型硅膠在無金屬離子環境下制造,所以表面不會殘留金屬離子也會減少拖尾。另外,硅膠表面盡可能減少自由的硅羥基,未與固定相鍵合的硅羥基盡可能形成螯合或聚合狀態,用來減少拖尾峰的產生。
2、當色譜柱經歷過高溫、強酸,或者一些極端使用方法導致固定相流失較多后,色譜柱中的自由硅羥基也會變得越來越多從而更加容易造成拖尾峰。
3、拖尾峰的形成也有可能是因為柱外的死體積較多,導致樣品在死體積處發生了滯留和擴散。
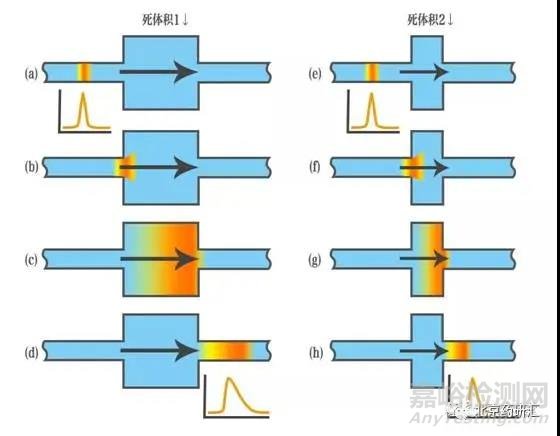
4、當樣品中出現了堿性化合物,堿性基團更容易與色譜柱中的硅羥基發生作用形成拖尾峰。
注:氣相色譜造成色譜峰拖尾的原因
前沿陡峭、后沿較前沿平緩的不對稱峰,稱為拖尾峰。氣相色譜中,常見的吸附色譜法(利用吸附劑表面對不同組分物理吸附性能的差別,而使之分離的色譜法稱為吸附色譜法),如果吸附等溫線為非線性,當進樣試樣量超過一定數量時就會出現拖尾峰;分配色譜法(利用固定液對不同組分分配性能的差別,而使之分離的色譜法稱為分配色譜法),如果載體表面具有活性作用點,試樣量超過柱負荷或進樣方法不當等,都會出現拖尾峰現象。什么原因會導致色譜峰拖尾?

引起色譜峰拖尾的原因比較復雜,如柱子兩端安裝不正確,沒有達到進樣口分流點和檢測器處尾吹點位置;或安裝好后又在接頭處斷裂;柱外死體積較大;尾吹氣流量小,樣品在柱內或系統內壁非線性吸附;汽化室污染等原因都容易造成拖尾。具體原因如下:
①進樣量過大;
②進樣器污染或汽化室中的襯管堵塞;
③襯管未脫活,造成待測物被吸附后逐步釋放;
④載氣流速過高;
⑤載氣系統漏氣;
⑥色譜柱安裝不正確;
⑦色譜柱嚴重流失或污染;
⑧柱溫太低或高于溶劑沸點溫度;
⑨汽化室死體積太大;
⑩進樣口汽化室溫度太低。
如何改善拖尾峰?
1 與化學有關的拖尾問題 ?
(1)流動相中,加入30ml的三乙胺(用于堿性化合物)或醋酸胺(用于酸性化合物), 未知化合物加醋酸三乙胺;
(2)如仍然拖尾,將三乙胺換為二甲基辛胺(或醋酸二甲基辛胺);
(3)降低進樣量至1μg。
2 與色譜柱有關的拖尾問題
(1)如柱頭處有強保留的樣品組分積聚,反相柱可用20倍柱體積的96%的二氯甲烷與4%甲醇,加1%氫氧化銨混合液沖洗;正向柱可用甲醇沖洗;
(2)使用保護柱。
3 與HPLC系統有關的峰拖尾和峰加寬
(1)進樣體積過大,(通常≤25μL);
(2)進樣閥與色譜柱及檢測器之間的管路體積過大
(3)檢測器流通池的體積過大。
當然氣相又有點區別
還有要是其他峰不拖尾只有主峰拖尾那就可能是過載或與物質本身性質有關
注:如何改善氣相色譜造成色譜峰拖尾
考慮到引起色譜峰拖尾的原因較復雜,可從以下幾方面來分析解決。

①進樣量檢查是否太大,減少進樣量,觀察色譜峰拖尾改善情況。
②進樣口檢查進樣針針尖碰到并破壞了襯管內的填充物,堵在柱頭,也會導致色譜峰拖尾,應從襯管中取出部分填充物、清理掉破碎物,或使用無填充的襯管。
③襯管脫活進樣口襯管上的活性位點可能吸附待測組分導致出現拖尾峰,并可能損失靈敏度和重現性。對于不分流進樣或分析極性很弱的化合物時,使用脫活襯管能盡量避免拖尾。當峰拖尾情況發生時,應及時更換襯管,尤其對于痕量分析,全新的襯管比清洗后并重新脫活的襯管在避免色譜峰拖尾上表現更有效。
④色譜柱溫度超出色譜柱承受上限的溫度會造成固定相和膜表面的加速損壞,這樣會造成色譜柱的過分流失、降低柱效(分離度),尤其在有泄漏或載氣中氧含量較高時,過度加熱會大大加速并永久地損壞色譜柱,這樣待分析活性組分的色譜峰就容易形成拖尾。
⑤色譜柱污染應切去色譜柱前端被污染的0.5~1m,必要時還需更換進樣口襯管、隔墊,清洗進樣口,嚴重時需要更換色譜柱。
⑥色譜柱位置在進樣口中的位置不正確,泄漏或柱端切割不平整,均會導致色譜峰前伸或拖尾,應用專用色譜柱切割刀將柱端切割得干凈而平整,重新按照安裝尺寸安裝色譜柱。
⑦進樣方式不分流進樣或柱上進樣時,溶劑效應顯著,應降低初始色譜柱溫度,這樣可使保留值增加、峰拖尾會減弱。
(4)案例分析
①襯管受污染和濃度過大引起的拖尾正己烷為溶劑配制有機氯農藥標準溶液,用GC-ECD檢測,各化合物色譜峰都有拖尾,觀察儀器EFC,參數正常,氣體無泄漏;檢查進樣墊,無泄漏;檢查襯管,襯管內壁有黑色污染物,更換成新襯管,同樣的色譜條件下進樣,色譜峰拖尾有所改善,響應也有所提高;檢查進樣量,與以往無差別;將溶液濃度從2.0μg/mL稀釋為0.1μg/mL,進樣,色譜峰拖尾消失,表明溶液濃度過大,亦易引起色譜峰拖尾。

②色譜柱污染引起的拖尾色譜柱使用較長時間后,因受污染、柱流失嚴重,柱效降低。如HP一5(30m×0.32mm×0.25μm)色譜柱,使用四年,進敵敵畏標準溶液(0.1μg/mL),峰拖尾情況嚴重,老化色譜柱與切割柱前端都無濟于事。
換成DB一5(30m×0.32mm×0.50μm)色譜柱,也使用四年,已污染,進敵敵畏標準溶液(0.1μg/mL),亦有拖尾現象;換成無污染的色譜柱DB一1701(30m×0.25mm×0.25μm),同樣的條件下,進敵敵畏標準溶液(0.1μg/mL),峰形尖銳、峰寬較窄,拖尾現象解決。
③基質標準溶液進樣,改善目標峰拖尾農藥噻螨酮分析一般采用HPLC方法。研究中我們發現采用GC—MS亦能分析,但是乙腈溶劑所配標準溶液進樣,噻螨酮出峰嚴重拖尾且響應很低。
當分別采用QuChERs方法凈化后的芒果空白和西葫蘆空白提取液配制標準溶液進樣時,能明顯改善噻螨酮色譜峰形和響應,當然也看到噻螨酮的西葫蘆基質標準色譜峰呈現一定的圓頭峰,可能跟不同基質下,噻螨酮在進樣口活性位點上的吸附解吸附速度有關。

來源:Internet


