您當前的位置:檢測資訊 > 法規標準
嘉峪檢測網 2020-11-11 16:49
合成多肽藥物質控及雜質譜研究
胡玉璽1 蔣煜1 韓天嬌2 何馳宇1
(1 國家食品藥品監督管理總局藥品審評中心,北京100038;2國家食品藥品監督管理總局食品藥品審核查驗中心,北京100061)
[摘要] 本文主要以相關指導原則和電子刊物為基礎,探討合成多肽藥質量控制項目設置的合理性,同時對多肽藥物雜質譜研究應關注的內容進行分析。
[關鍵詞] 合成多肽藥;質量標準;雜質譜
[中圖分類號] R95 [文獻標志碼] C [文章編號]1003-3734(2018)05-0502-07
Study on quality control and related substances of synthetic peptide drugs
Hu Yuxi1 Jiang Yu1 Han Tianjiao2
(1 Center for Drug Evaluation, China Food and Drug Administration, Beijing 100038, China;2 Center for Food and Drug Inspection, China Food and Drug Administration, Beijing 100061, China)
[abstract] Referencing to relevant guidelines and electronic journals, this paper mainly discusses the scientific justification of specifications of synthetic peptide drugs,as well as the impurity analysis during synthetic peptide development.
[Key words] synthetic peptide drugs;quality standard;related substances
[概述] 合成多肽藥物可視為界于有機小分子和蛋白質大分子之間的一類特殊藥物。在《制備工藝和過程控制對合成多肽藥物有關物質的影響》[1]中已對合成多肽藥物制備工藝部分進行了論述,本文主要就合成多肽藥物質控項目設置的合理性和雜質譜研究的全面性進行探討。
1、相關技術指導原則
合成多肽作為一類特殊的藥物,其氨基酸序列,潛在的二級和三級結構,以及制備工藝和質控要求等,與經典小分子化學藥物相比有明顯差別,具有多樣性和復雜性,也可能是這個原因,國內外指導文件很少涉及此類品種。FDA于1994年發布了“Guidance for Industry for the Submission of Chemistry,Manufacturing,and Controls Information for Synthetic Peptide Substances” [2],國家食品藥品監督管理總局于2007年發布了“合成多肽藥物藥學研究技術指導原則”[3],兩個指導原則對合成多肽藥物的制備工藝、結構確證、質量研究與質量標準和穩定性研究等方面提出了相應的技術要求,在當時對多肽藥物的研究和開發起到了積極的指導作用。但隨著藥品開發和質控理念的提升,未見國內外相關技術指導原則進行相應更新。如何結合最新藥品研發理念,合理的開發并控制多肽類藥物的質量具有重要意義。
2、多肽原料藥質控概況
目前,國內外藥典收載了十幾個合成多肽藥物,質量標準中規定的控制項目主要包括:外觀、比旋度、鑒別、酸度、溶液澄清度與顏色、氨基酸組成、殘留溶劑(醋酸、三氟乙酸)、水分、有關物質、微生物限度、含量等。美國藥典委員會多肽專家小組于2014年發表了相關文章論述了多肽藥物質控標準及相應分析方法,見表1[4]。質控項目中氨基酸序列、肽譜、氨基酸組成等為多肽類藥物特有質控項目,也是多肽藥物的常規檢查項目。與經典小分子化藥相比,合成多肽藥物在鑒別和有關物質檢查項目存在一定特殊性。
表1 多肽藥物的質量控制項目和分析方法

a:批次放行時需要檢測的項目;HPLC:高效液相色譜法;MS:質譜;NMR:核磁共振;FTIR:傅里葉變換紅外光譜
2.1 鑒別
藥典收載的合成多肽類原料藥鑒別項目多為以下方法:高效液相法(HPLC法,色譜條件與含量測定項下方法相同)、紅外法、雙縮脲顯色反應等,對于多肽類藥物來說,上述鑒別項目專屬性較差,需考慮增加專屬性更強的檢測手段進行鑒別考察,如肽譜、核磁或質譜等鑒別方法。如現行版美國藥典中,收載的去氨加壓素原料藥,鑒別項目采用質譜檢測;現行版歐洲藥典中,收載的布舍瑞林和戈舍瑞林原料藥鑒別項目采用核磁檢測。采用質譜法鑒別時,其結果應在理論值的±1.0質量單位內。
核磁共振光譜鑒別適用于氨基酸數目較少的多肽藥物,對于超過10個氨基酸殘基的多肽,核磁共振光譜數據分析會受到一定的挑戰,可以選擇使用肽譜進行鑒別研究。
美國藥典委員會多肽專家小組建議如選擇HPLC定位方法作為鑒別方法,應對樣品和標準品主峰的保留時間進行比較,并要求標準品和樣品等劑量混合進樣時,能得到單峰。
2.2 有關物質
合成多肽藥物的雜質譜復雜,包含由起始物料引入的雜質、工藝相關的雜質和降解產生的雜質等,為實現有效檢出各潛在雜質,通常需要采用多種不同原理的檢測方法如反相HPLC法,離子交換HPLC法,分子排阻HPLC法等。如某進口39肽藥物,原料藥中控手段采用反相HPLC法控制13個已知雜質,最大未知單個雜質和總雜質,離子交換HPLC法控制2個特定雜質,分子排阻HPLC法控制二聚體等聚合物雜質。
現行版歐洲藥典通則中提出的多肽原料藥雜質限度要求比ICH Q3A指導原則的要求更寬泛,報告閾值、鑒定閾值和界定閾值分別為0.1%、0.5%和1.0%[5],主要依據可能是與傳統小分子藥物的雜質相比,多肽類雜質引發毒理學問題的風險較低。美國藥典委員會多肽專家小組對多肽藥物的雜質限度存在其他要求,認為不同的多肽藥物的活性存在較大差異,不同的雜質可能同樣存在活性的差別,同時多肽類藥物多為注射劑,風險相對較高。因此,該小組認為多肽藥物雜質控制限度的制定應該結合特定品種進行具體分析。
3、雜質譜研究
如《制備工藝和過程控制對合成多肽藥物有關物質的影響》[1]所述,多肽藥物雜質譜復雜,應結合起始物料情況、制備工藝和穩定性考察結果對多肽藥物進行系統的雜質譜分析。目前,國內申請人向CFDA遞交的合成多肽藥物雜質譜研究資料要么不夠充分,考察雜質較少,未對由起始物料引入的雜質、工藝雜質和降解雜質等進行系統的考察;要么考察數十個雜質,堆積“龐大”的雜質譜研究資料,將合成、降解引入的雜質與其他雜質混為一體,缺乏針對性分析和研究。以某28肽藥物為例,共考察了65個潛在雜質,所研究的雜質譜中有23個缺失10至16個氨基酸殘基的缺失肽雜質,暫且不考慮上述雜質在氨基酸偶聯過程中生成的可能性,該產品制備工藝最后精制過程中有柱層析提純步驟,缺失氨基酸數目較多的缺失肽雜質性質與目標多肽性質相差較大,即使在氨基酸偶聯過程中能產生上述雜質,在柱層析過程中也能有效除去,引入終產品的可能性很小,提供的終產品檢測報告中也證明了上述雜質在終產品中均不存在。另外,本品雜質譜研究中,對潛在易水解和易消旋的氨基酸引入的雜質均未進行針對性研究,因此盡管考察雜質數量很多,雜質譜研究仍存在較大缺陷。綜上,對組成氨基酸的性質、整個制備過程和潛在的降解途徑的理解,對合成多肽的雜質譜研究尤為重要。
美國藥典委員會多肽專家小組發表的文章中,涉及了合成多肽藥物雜質譜研究內容和考察方法,見表2和表3[3]。《制備工藝和過程控制對合成多肽藥物有關物質的影響》一文中也對可能由起始物料和制備過程中引入的缺失肽、錯結肽等雜質進行了討論[1]。本文主要針對差向肽雜質、鏈端雜質、側鏈雜質、氧化雜質等進行討論。
3.1 差向肽雜質
差向肽雜質為氨基酸序列中含一個或多個非預期手性構型的氨基酸殘基所形成的肽鏈,差向肽雜質可能形成于原料氨基酸中的光學異構體,因此需要對原料進行嚴格控制;也可能形成于肽鏈合成過程中差向異構化。目前已知的差向異構化機理包括:直接烯醇化和生成惡唑酮(氮雜內酯)。機理見圖1。在Fmoc保護策略的固相合成工藝中,由于避免了活化肽鏈端羧基,差向肽主要形成機理為氨基酸偶聯過程中直接烯醇化,在氨基酸偶聯過程中所使用的催化劑等堿性試劑具有催化脫氫作用,形成碳負離子,而部分氨基酸的側鏈基團具有穩定該碳負離子的作用,因此在偶聯過程中易產生相應的差向肽雜質,如半胱氨酸(Cys),組氨酸(His)。

圖1 差向肽雜質形成機理
相對于其他類型雜質來說,差向肽雜質鑒定和分離均有較大難度。尤其對于含有氨基酸數目較多的多肽藥物,對每一個手性氨基酸均利用合成相應的差向肽雜質對照品,使用HPLC出峰時間對比鑒定的方法工作量巨大。可采用氯化氘[DCl] /氘代水[D2O]水解衍生后,采用手性氣相色譜-質譜(GC-MS)來測定每個氨基酸手性異構體的含量,鑒別出易產生消旋的氨基酸,進一步制備出相應的差向肽雜質對照品進行HPLC出峰時間對比鑒定,證明擬定分析方法可以有效檢出易產生的差向肽雜質。
3.2 鏈末端雜質
鏈末端雜質包括兩大類:一類是N端酰化或C端脫酰胺化產生的雜質,該類雜質較易得到雜質對照品,研究難度不大。另一類雜質是由鏈末端活性基團“反咬”所形成的雜質,如N端的氮原子對第2個與第3個氨基酸殘基之間酰胺鍵中的羰基發生親核進攻,隨后導致上述兩個氨基酸殘基發生截斷,生成二酮哌嗪類物質,肽鏈生成少兩個氨基酸的缺失肽雜質。當N端為位阻較小的脯氨酸,甘氨酸殘基時,有利于上述副反應的發生。另一方面,如N端氨基酸殘基為谷氨酰胺時,則焦谷氨酸生成反應可能性很大。與上述反應機理類似,即N端氮原子進攻谷氨酰胺側鏈羰基碳后生成焦谷氨酸多肽類似物。N端殘基為天冬酰胺時也可發生這一轉化過程。

圖2 二酮哌嗪和焦谷氨酸降解機理
3.3 側鏈雜質
側鏈雜質主要可分為2類:側鏈保護基團引入的雜質和氨基酸側鏈本身具有反應活性引入的雜質。在多肽合成過程中,側鏈保護基團在酸解脫除過程中,可能存在脫除不完全,同時形成的碳正離子可能與肽鏈中潛在的親核位點偶聯,形成其他雜質。此類雜質一般會表現出與目標多肽分子量、極性等方面的差異,采用適當的手段可以充分說明。
由氨基酸側鏈本身具有反應活性引入的雜質的分析需建立在對所組成氨基酸性質充分理解的基礎上,較常見的是由谷氨酰胺、天冬酰胺側鏈酰胺鍵水解產生的降解雜質。上述氨基酸可以進一步發生如下副反應,由天冬酰胺水解產生的天冬氨酸,或多肽序列中含有的天冬氨酸,則易生成琥珀酰亞胺結構,通過C端酰胺基氮原子對天冬酰胺或天冬氨酸殘基的γ-羰基發生親核進攻,形成五元琥珀酰亞胺環的反應。弱酸性條件有利于上述成環反應,生成的環狀結構在生理條件和堿性條件下均不穩定,可發生消旋化。琥珀酰亞胺環的消旋化和水解可生成由L-天冬氨酸、D-天冬氨酸、L-異天冬氨酸和D-異天冬氨酸組成的混合物,因此可使原天冬酰胺殘基發生脫酰胺基化和消旋化,或可使原天冬氨酸殘基發生異構化和消旋化生成系列雜質。如肽鏈中含有谷氨酰胺或谷氨酸時,也可發生類似的副反應。

圖3 琥珀酰亞胺降解機理
精氨酸側鏈可以發生氨基化,反應生成一級胺,轉化為鳥氨酸殘基的副反應。該鳥氨酸殘基含一個未受保護的氨基,因此可與其他氨基酸殘基發生偶聯,進一步生成其他的雜質。
半胱氨酸中碳硫鍵在氫氧根離子催化下可發生β-消除的副反應,該副反應將生成兩種新殘基(即脫氫丙氨酸殘基和硫代半胱氨酸殘基)。脫氫丙氨酸可與合成體系中含有的賴氨酸或其他含有活性氨基化合物反應,發生賴氨酸-丙氨酸交聯,生成聚集體雜質,硫代半胱氨酸則可進一步降解為含巰基的產物,相關降解途徑可參見圖4。絲氨酸和蘇氨酸殘基也可能發生β-消除副反應。

圖4β-消除降解機理
3.4 氧化/還原雜質
固相合成過程中,特定氨基酸殘基可能發生氧化或還原反應。已在卡貝縮宮素和依來多辛中分別發現了因半胱氨酸和甲硫氨酸殘基氧化而形成的雜質。上述雜質相對于目標序列多肽的質量差異為16、32和48 Da,為不同氧化階段產生的雜質,即亞砜、砜和磺酸。組氨酸和賴氨酸也易發生氧化,分別生成2-氧代-組氨酸和氨基己二酸,貯存過程中長時間光照或暴露在空氣中均可導致生成該氧化雜質。此外,色氨酸側鏈在酸性條件下的反應活性較高,可發生氧化生成系列雜質,還可發生還原反應。易發生氧化和還原的氨基酸殘基的總結情況可參見圖5。
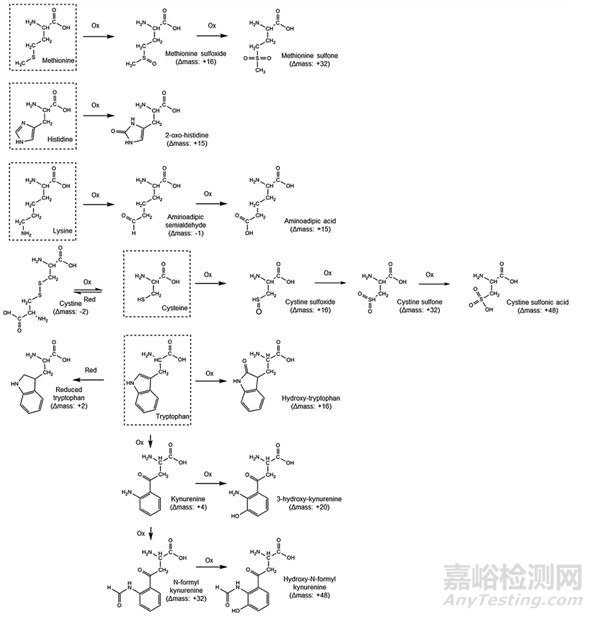
圖5 氨基酸氧化/還原相關雜質
3.5多聚體雜質
多肽藥物在純化和放置過程中,由于端基具有反應活性或二硫鍵的改變等,可能生成一定量的多聚體雜質。如去氨加壓素中,發現并鑒定出的多聚體雜質有6種之多。多聚體雜質一般可使用分子排阻色譜法進行有效的檢測。
[小結] 合成多肽藥物的開發需要建立在對多肽藥物充分理解的基礎上,建立全面的質控策略;雜質譜研究應對起始物料引入的雜質、制備工藝中生成的雜質、降解過程產生的雜質進行充分考察,保證雜質譜研究的全面性。
參考文獻:
[1] 胡玉璽,蔣煜,韓天驕. 制備工藝和過程控制對合成多肽藥物有關物質的影響[J].中國新藥雜志,2017,26(18):2143-2148.
[2] U.S. FDA Guidance for Industry for the Submission of Chemistry,Manufacturing,and Controls Information for Synthetic Peptide Substances[S].1994.
[3] 國家食品藥品監督管理總局. 合成多肽藥物藥學研究技術指導原則[S].2007.
[4] EGGEN I. Control strategies for synthetic therapeutic peptide APIs Part I:analytical considerations[J]. Pharma Technol, 2014,27(3):16-21.
[5] Ph. Eur., General Guidance 2034, Substances for Pharmaceutical Use[S].2015.

來源:Internet


