您當(dāng)前的位置:檢測資訊 > 科研開發(fā)
嘉峪檢測網(wǎng) 2019-12-03 11:30
張素才 1, 姚大林 1, 孫云霞 1, 孫濤 2
1. 北京昭衍新藥研究中心股份有限公司, 生物制品安全性評價北京市重點實驗室, 北京 100176;
2. 國家藥品監(jiān)督管理局藥品審評中心, 北京 100022
摘要:
致癌性研究是藥物非臨床安全性評價的重要內(nèi)容之一,致癌性試驗實施的復(fù)雜程度遠遠超出指導(dǎo)原則的要求。本文對美國FDA在2014-2018年5年期間批準的213個新藥進行了梳理和分析,并結(jié)合ICHS1要求、文獻報道和實際工作經(jīng)驗,從致癌性試驗的必要性、致癌性試驗結(jié)果提交時間、生物制品致癌性試驗的決策、致癌性試驗的試驗類型選擇、致癌性試驗的劑量設(shè)計等幾方面提出意見和建議,力求為國內(nèi)同行、新藥研發(fā)企業(yè)和審評機構(gòu)提供參考。
1 前言
2017年6月, 國家藥品監(jiān)督管理局(原國家食品藥品監(jiān)督管理總局)正式加入國際人用藥品注冊技術(shù)要求協(xié)調(diào)會(ICH), 成為全球第8個監(jiān)管機構(gòu)成員, 標志著我國藥品標準在國際合作領(lǐng)域邁出重要的一步。而根據(jù)ICH發(fā)布的協(xié)會章程規(guī)定, 監(jiān)管機構(gòu)成員要逐步實施ICH技術(shù)指導(dǎo)原則, 因此, 藥物研發(fā)的所有參與者(申請人、合同研究機構(gòu)、監(jiān)管機構(gòu)等)均將以ICH指導(dǎo)原則作為行動指南, 實施初期將面臨較大的挑戰(zhàn)。
盡管科學(xué)界對動物致癌性試驗, 尤其是嚙齒類動物2年致癌性試驗的存在價值有很多的爭議和討論, 但是在監(jiān)管層面達成一致意見之前, 國內(nèi)的新藥申報企業(yè)和研究機構(gòu)仍需按照ICH S1的國際規(guī)范全面實施。ICH S1指導(dǎo)原則自從1995年頒布后, 在不斷完善, 目前包含三份文件[1-3], 分別是1995年頒布實施的"Guideline on the Need for Carcinogenicity Studies of Pharmaceuticals"(ICH S1A:藥物致癌性試驗必要性的指導(dǎo)原則)、1997年的"Testing for Carcinogenicity of Pharmaceuticals" (S1B:藥物的致癌性試驗)和"Dose Selection for Carcinogenicity Studies of Pharmaceuticals"[S1C (R2):藥物致癌性試驗的劑量選擇]。貫徹實施ICH指導(dǎo)原則的前提在于深入了解制定規(guī)范的科學(xué)基礎(chǔ), 知其然并知其所以然; 結(jié)合實際案例的指導(dǎo)原則學(xué)習(xí), 將會起到事半功倍的效果。美國食品藥品監(jiān)督管理局(Food and Drug Administration, FDA), 藥品評價與研究中心(Center for Drug Evaluation and Research, CDER)近5年內(nèi)(2014- 2018年)共批準了153個新藥申請(New Drug Application, NDA)和60個生物制品許可申請(Biologic Licensing Application, BLA)[4], 本文將對這213個新藥進行梳理分析, 從藥物致癌性試驗的必要性、生物制品致癌性試驗的決策、試驗類型的選擇以及劑量選擇等幾方面提出一些意見和建議, 以期幫助藥物研發(fā)參與者加深對ICH S1的理解, 為新藥研發(fā)和申報工作提供參考。
2 致癌性試驗的必要性
2.1 ICH S1A的要求
致癌性試驗的目的在于發(fā)現(xiàn)和識別藥物對動物的潛在致瘤性(包括良性、惡性), 繼而評價其對人體的相關(guān)風(fēng)險。評估致癌性試驗必要性的最基本考慮是臨床用藥周期長度以及其他安全性研究所發(fā)現(xiàn)的相關(guān)結(jié)果; 其他因素也應(yīng)被考慮, 如患者人群數(shù)量、性別、潛在致癌性的預(yù)評估、系統(tǒng)暴露程度、與內(nèi)源性物質(zhì)的異同、與臨床研究進程相對應(yīng)的試驗時間安排等。
總體來說, 需要進行致癌性試驗的情形包括:病人長期應(yīng)用的藥物(關(guān)于用藥周期在下文討論), 或頻繁的間歇性用藥以致總的暴露時間與連續(xù)用藥類似的藥物; 可能導(dǎo)致延長暴露時間的給藥藥物系統(tǒng); 同類產(chǎn)品先前已證明有與人相關(guān)的致癌性; 構(gòu)效關(guān)系提示有致癌風(fēng)險的; 在亞慢性或慢性重復(fù)給藥毒性試驗中有不典型增生或癌前病變的; 在組織內(nèi)長期滯留的母體化合物或其代謝產(chǎn)物導(dǎo)致局部組織增生或其它病理生理變化的; 一些抗腫瘤藥物, 擬用于非帶瘤病人的輔助治療或非腫瘤適應(yīng)癥長期使用時也需考慮進行致癌性試驗。
不需要進行致癌性試驗的情形主要包括:非經(jīng)常使用或短期暴露的藥物(如麻醉藥和放射性同位素標記造影劑); 明確有遺傳毒性的化合物而臨床中仍需長期使用的, 不一定需要進行長期致癌性試驗, 但有必要進行長期毒性試驗(長達1年)以觀察其有無早期致癌作用; 擬定治療人群的預(yù)期壽命較短(如2~3年之內(nèi))的藥物; 系統(tǒng)暴露量小的局部用藥; 對于改鹽、改酸根或堿基的藥物, 經(jīng)藥代動力學(xué)、藥效動力學(xué)和毒性等方面評估藥物暴露量和毒性無明顯變化時可不需進行致癌性試驗。
2.2 美國FDA批準的213個新藥
從表 1和圖 1可見, 在上述已批準的213個新藥中, 申請上市前完成動物致癌性試驗比例較高的幾類藥物分別是精神類藥物, 肺部、變態(tài)反應(yīng)和風(fēng)濕病藥物, 以及代謝和內(nèi)分泌類藥物, 這也是與大家已有的觀念相同(即臨床長期用藥)。以下將從臨床用藥周期、未進行致癌性試驗的原因分類、致癌性試驗結(jié)果的提交時間、生物制品致癌性試驗的決策和不要求而實際進行致癌性試驗的情形等5方面進行討論分析。
表 1 美國FDA在2014年-2018年期間批準的213項新藥分類
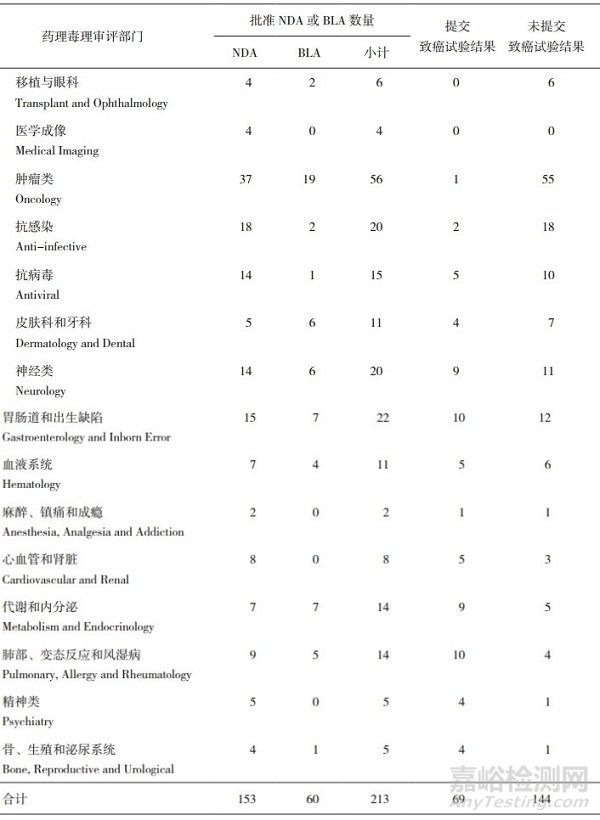
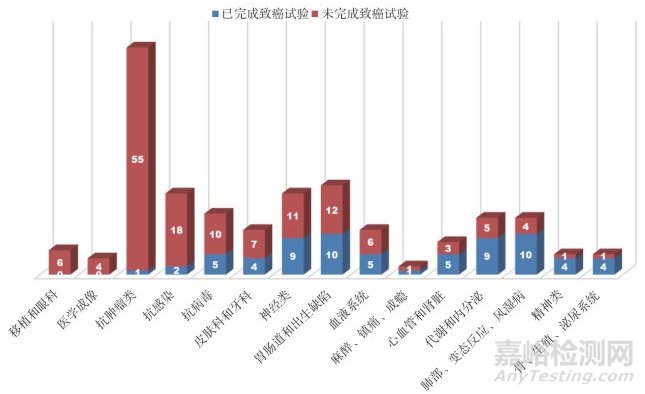
圖 1 2014-2018年美國FDA批準新藥的致癌性試驗開展情況
2.2.1 關(guān)于臨床用藥周期
FDA要求, 一般藥物使用3個月或更長時間, 需要進行動物致癌性試驗。所以在美國大部分長期使用的藥物在廣泛應(yīng)用于人體之前, 都已進行了動物致癌性研究。在歐洲, "歐共體藥品管理條例"規(guī)定需要進行致癌性試驗的情況, 包括長期應(yīng)用的藥物, 即至少6個月的連續(xù)用藥或頻繁的間歇性用藥以致總的暴露量與前者相似的藥物。日本1990年"藥物毒性研究指導(dǎo)原則手冊"規(guī)定:如果臨床預(yù)期連續(xù)用藥6個月或更長時間, 則需要進行致癌性試驗。盡管連續(xù)用藥少于6個月, 如果存在潛在致癌性因素, 也可能需要進行致癌性試驗。由此可見, 臨床用藥周期長短是判定致癌性試驗必要性的首要考慮因素, 連續(xù)用藥6個月是一個共識。2015年FDA批準的艾沙康唑(Isavuconazonium, NDA 207500), 用于18歲以上的侵入性曲霉病和毛霉菌病; 申請人引用ICH S1A中的連續(xù)用藥3個月并依據(jù)其他唑類藥物已有動物致癌性的報道, 申請豁免動物致癌性試驗; 而FDA審評員的觀點是大多數(shù)連續(xù)使用3個月的藥物, 用藥周期多會達到6個月, 并且要求在藥品說明書(Label)中增加"該藥未進行2年致癌性試驗, 其他唑類藥物的小鼠和大鼠致癌性試驗中可見肝細胞腺瘤和肝癌", 并須在上市后完成嚙齒類動物2年致癌性研究[5]。因此, 在美國FDA的上市后要求(Postmarket Requirements)網(wǎng)頁中顯示2年小鼠致癌性試驗和2年大鼠致癌性試驗已在2019年5月完成。
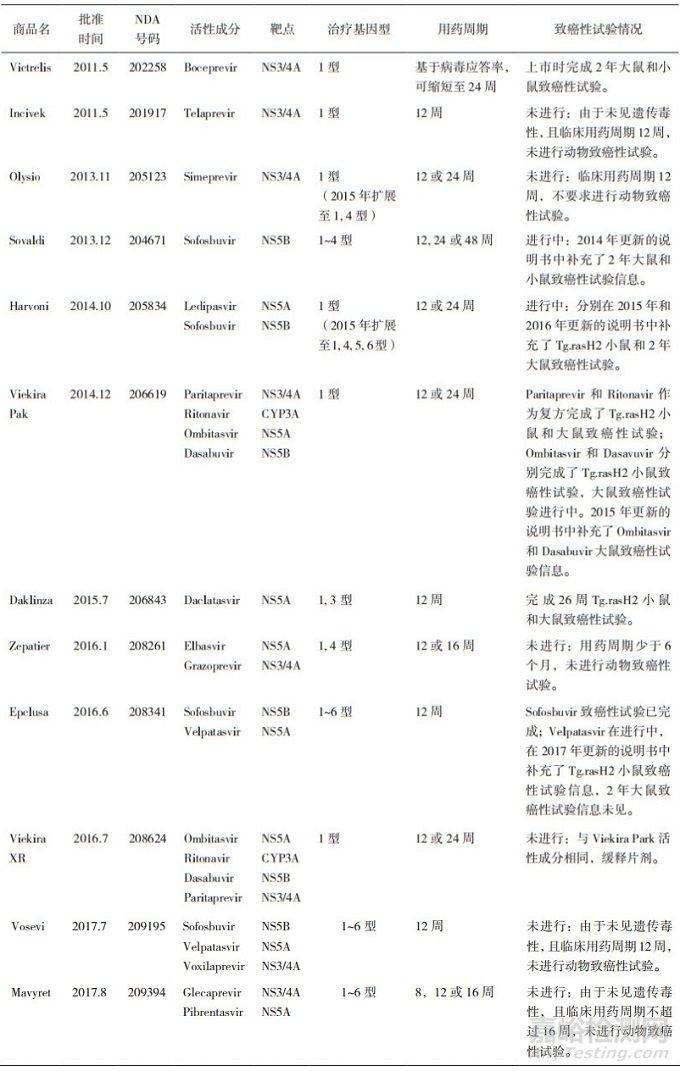
對于用藥周期的判定也不是一成不變的, 隨著科學(xué)界對疾病和藥物作用機理認識程度的深入, 用藥周期也會有所改變; 用于治療慢性丙型肝炎(CHC)的直接抗病毒藥(DAA)就是其中的典型代表, 隨著對丙肝病毒(HCV)復(fù)制、生命周期、病毒基因型及病毒蛋白晶型結(jié)構(gòu)認識的深入, 對于該類藥物在動物致癌性試驗的認識上也在發(fā)生著一些改變。自2011年美國FDA批準第一個非結(jié)構(gòu)蛋白(NS3/4A)酶抑制劑至2018年底, FDA共批準了5個單藥和7個復(fù)方藥用于治療CHC, 其中含有16個新分子實體(New Molecular Entity, NME)。從表 2可見, Viekira XR因與Viekira Pak具有相同的活性成分而除外, FDA批準的11個DAA藥物中, 6/11個由于未見遺傳毒性, 且臨床用藥周期少于6個月, 而未進行動物致癌性試驗; 其余5/11個都開展了致癌性試驗, 但僅2/5個在批準上市時提交了完整的致癌性試驗數(shù)據(jù), 3/5個在批準上市時未提交或提交部分致癌性試驗結(jié)果, 但在上市后完成了補充試驗。新一代NS3/4A絲氨酸蛋白酶抑制劑單用、復(fù)方, 或與干擾素和利巴韋林聯(lián)用, 臨床療程可縮短至12周, 甚至8周, 且單一療程就能達到很高的治愈率, FDA接受其不進行動物致癌性試驗而批準藥物上市[6]。
表 2 美國FDA批準的不同靶點DAA藥物致癌性試驗信息歸納
2.2.2 藥物未進行致癌性試驗的原因分類
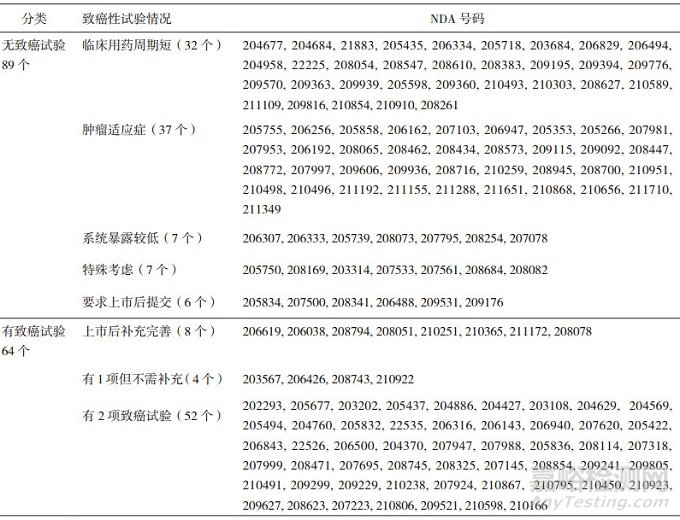
將遞交NDA時未進行或尚未完成動物致癌性試驗的項目簡稱為無致癌性試驗, 將完成至少1項動物致癌性試驗的項目簡稱為有致癌性試驗; 從表 3可見, 在過去5年期間, FDA批準的153個NDA項目中, 無致癌性試驗的是89個, 有致癌性試驗的是64個。無致癌性試驗的最常見理由是藥物為腫瘤適應(yīng)癥(約42%)或臨床用藥周期較短(約36%); 還有14/89個是因為系統(tǒng)暴露較低或其他原因(表 3中簡稱為特殊考慮的); 6/89個藥物是獲得推遲提交致癌性試驗結(jié)果的許可(上市后提交), 將在下文另行討論。
表 3 美國FDA批準的153個NDA關(guān)于動物致癌性試驗的分類
根據(jù)ICH S1的要求, 系統(tǒng)暴露量小的局部用藥不需要以經(jīng)口給藥途徑來評價其對內(nèi)臟器官的潛在致癌作用。從表 3可見, 非那沙星耳懸液(Finafloxacin Otic Suspension, NDA206307)[7]、Deoxycholic Acid(NDA206333)[8]和Lifitegrast Ophthalmic Solution(NDA208073)[9], 3個藥物分別用于治療急性外耳道炎、成人下頜中重度脂肪堆積和干眼癥; Vyzulta(Latanoprostene Bunod Ophthalmic Solution, NDA207795)[10]和Rhopressa (Netarsudil Ophthalmic Solution, NDA208254)[11]均用于治療開角型青光眼; 上述5個藥物均屬于局部用藥。而Patiromer(NDA205739)[12]和Sodium Zirconium Cyclosilicate(NDA207078)[13], 盡管均屬于口服給藥用于高鉀血癥的治療, 但屬于鉀離子結(jié)合樹脂或鉀黏結(jié)劑, 系統(tǒng)暴露較低, 故未進行動物致癌性研究。
按照臨床用藥周期, 需要進行動物致癌性試驗但沒有進行, 且不屬于腫瘤適應(yīng)癥或系統(tǒng)暴露較低的藥物有7個:Cholbam(Cholic acid, NDA205750)[14], 是膽汁酸的替代治療, 適應(yīng)癥是罕見膽汁酸合成障礙, 未進行非臨床安全性評價動物試驗。尿苷三乙酸酯(Uridine Triacetate, NDA208169)[15], 屬于嘧啶模擬物, 用于治療遺傳性乳清酸尿癥, 尿苷三乙酸酯在體內(nèi)快速轉(zhuǎn)化為尿苷, 而外源性尿苷已被用于各種適應(yīng)癥, 包括糖尿病腎病、線粒體和神經(jīng)代謝紊亂, 以及5-氟尿嘧啶毒性, 安全性風(fēng)險較低。德谷胰島素(Insulin Degludec, NDA203314)[16], 屬于胰島素類似物, 進行了大鼠重復(fù)給藥52周的毒性試驗, 動物未見藥物相關(guān)的增生性改變或良性/惡性腫瘤發(fā)生。月桂酰阿立哌唑(Aripiprazole Lauroxil, NDA207533)[17], 用于精神分裂癥的治療, 在本品的任何非臨床研究中均未見癌前病變, 且已進行口服阿立哌唑的2年小鼠和大鼠致癌性試驗, 故未再進行月桂酰阿立哌唑的動物致癌性試驗, 但需要在說明書中體現(xiàn)阿立哌唑的動物致癌性試驗信息。Genvoya (NDA207561)[18], 為治療HIV的固定劑量復(fù)方制劑, 其中新分子體替諾福韋艾拉酚胺(Tenofovir Alafenamide, TAF)是替諾福韋(Tenofovir, TFV)的前藥, 與TFV的另一個前藥富馬酸替諾福韋酯(Tenofovir Disoproxil Fumarate, TDF)相比, TAF在血漿中更穩(wěn)定, TAF進入外周單核細胞, 在細胞內(nèi)代謝為TFV, 并磷酸化為活性代謝產(chǎn)物替諾福韋二磷酸(TFV-DP), 給予TAF可導(dǎo)致HIV靶細胞中TFV-DP水平升高, 同時降低TFV的循環(huán)水平, 有望降低TFV的脫靶效應(yīng), 提高安全性。由于TAF在小鼠和大鼠體內(nèi)可快速轉(zhuǎn)化為TFV, 因此, 未進行TAF的致癌性研究。Emflaza(Deflazacort, NDA208684)[19], 屬于皮質(zhì)類固醇藥物前體, 用于治療杜氏肌營養(yǎng)不良, 由于嚙齒動物不能預(yù)測免疫抑制劑類藥物(包括糖皮質(zhì)激素)與人體相關(guān)的腫瘤類型, FDA同意豁免2年大鼠致癌性試驗。Deutetrabenazine(NDA208082)[20], 美國FDA批準的首個氘代藥物, 是四苯喹嗪(Tetrabenazine)的氘代產(chǎn)物, 藥代動力學(xué)特征在氘代以后得到改善, 半衰期明顯延長, 從而可以使用更低的治療劑量; 由于在Tetrabenazine給予p53+/-小鼠26周的致癌性試驗中未見腫瘤發(fā)生率增加, 故針對本品未進行致癌性研究。
2.2.3 致癌性試驗結(jié)果的提交時間
當(dāng)需要進行致癌性試驗時, FDA要求申請人在遞交NDA之前或遞交NDA同時提供致癌性試驗研究結(jié)果。若對患者人群存在特殊擔(dān)憂, 在進行大樣本臨床試驗之前需完成嚙齒類動物的致癌性試驗[21]。對于開發(fā)用于治療某些尚無有效治療手段的嚴重疾病的藥物, 或用于老齡病人群體的藥物, 申請上市前可不必完成動物致癌性試驗, 但要求在上市后伴隨4期臨床進行致癌性試驗。
從藥物開發(fā)成本的角度考慮, 為獲得最大收益, 申請人也希望在上市后提交或補充完善動物致癌性試驗資料; 過去5年期間, 共有14個藥物存在這樣的情況, 其中的3個治療丙肝的藥物在前文已討論, 從表 4所列的11個藥物可見:6/11個是神經(jīng)類藥物, 1/11個是針對囊性纖維化的藥物, 還有1/11個是針對HIV感染的藥物。FDA會根據(jù)每個藥物的自身特點, 權(quán)衡臨床獲益與風(fēng)險管控, 特別是對適應(yīng)癥較為嚴重、醫(yī)療需求尚未得到滿足的新藥, 會批準其上市后進行或補充完善致癌性試驗資料。
表 4 上市后提交或上市后補充完善的案例

2.2.4 生物制品致癌性試驗的決策
ICH S1中針對生物制品(大分子藥物)的主要要求:對于一些經(jīng)化學(xué)合成、由動物或人體來源提取純化或通過生物技術(shù)方法生產(chǎn)的內(nèi)源性肽類或蛋白質(zhì)類物質(zhì)及其類似物, 所產(chǎn)生的生物作用與天然產(chǎn)物明顯不同, 或經(jīng)過修飾導(dǎo)致產(chǎn)品結(jié)構(gòu)與天然產(chǎn)物相比有明顯變化, 或在人體局部或全身的濃度明顯增加(即達到藥理學(xué)水平)時應(yīng)考慮進行致癌性試驗; 而作為替代性治療的內(nèi)源性物質(zhì), 且濃度在生理水平時可不需進行致癌性試驗[1]。
對于生物制品(大分子藥物)是否進行致癌性試驗的早期決策路線是過于簡單的, 主要兩點考慮, 其一是臨床用藥周期, 其二是嚙齒動物致癌性研究的可行性, 包括是否在動物體內(nèi)產(chǎn)生藥理活性, 以及是否產(chǎn)生中和抗體。但針對生物制品的決策路線, 如今新的傾向[22]是除了臨床用藥周期外, 還要考慮風(fēng)險擔(dān)憂, 包括是否屬于生長因子類、是否存在免疫抑制作用等, 如果沒有這些方面的擔(dān)憂, 只需書面說明不進行動物致癌性試驗的理由; 如果存在以上擔(dān)憂, 就需進一步采取證據(jù)權(quán)重(Weight of Evidence)分析, 分析內(nèi)容主要包括以下方面:(1)靶點作用是否與腫瘤相關(guān); (2)是否可采用基因敲除小鼠來說明致癌性特征; (3)是否可從腫瘤促進模型中獲得信息; (4)是否可獲得體外細胞增殖試驗; (5)人體基因變異與靶點調(diào)控作用的相似性; (6)進行2年嚙齒動物致癌性試驗是否可降低風(fēng)險; (7)患者的風(fēng)險和獲益考慮; (8)是否可通過說明書或上市后監(jiān)管來控制風(fēng)險。申請人應(yīng)根據(jù)以上幾點提出書面依據(jù)。IL-17A單抗(Ixekizumab, BLA125521)[23]關(guān)于致癌性試驗的權(quán)重分析方法就是一個很好的參考, 簡要來說:從物質(zhì)基礎(chǔ)上來看, 該單抗是大分子蛋白, 不進入細胞核, 不與DNA相互作用。前期毒理試驗結(jié)果, 在非人靈長類毒性試驗中, 對淋巴細胞亞群、NK細胞功能均無改變, 不影響動物的T細胞依賴性抗體反應(yīng)(TDAR), 淋巴器官無病變。從靶點調(diào)節(jié)與腫瘤的相關(guān)性上來看, 沒有確切結(jié)論, IL-17A有明確促炎作用, 而慢性炎癥與腫瘤的相關(guān)性也早已得到證實, 本品是IL-17A拮抗劑, 有一定的抑制炎癥作用; 但是也有研究顯示IL-17A有抗腫瘤作用; 即有研究表明, 采用IL17缺陷小鼠可見腫瘤生長加快或轉(zhuǎn)移現(xiàn)象。體外試驗研究, IL-17A可促進細胞增殖。臨床報道顯示, IL17缺陷個體易發(fā)口腔念珠菌感染。本品在正常嚙齒動物體內(nèi)不具有藥理學(xué)活性。從風(fēng)險獲益角度考慮, 本品用于中重度斑塊性銀屑病和活動性銀屑病關(guān)節(jié)炎。總體來說, IL17的抑制可以為腫瘤生長創(chuàng)造不利環(huán)境, 可通過上市后監(jiān)測提供最終風(fēng)險評估。審評人員最終同意豁免嚙齒動物致癌性試驗研究。
過去5年, FDA批準的60個大分子藥物中, 絕大多數(shù)(55/60個)未進行嚙齒動物致癌性試驗, 僅有5/60個藥物進行了1項或2項動物致癌性試驗(詳見表 5)。2個GLP-1受體激動劑(Dulaglutide和Semaglutide)[24-25], 與其他已上市的同靶點藥物相似, 具有甲狀腺腫瘤的擔(dān)憂, 且2個藥物在嚙齒動物體內(nèi)具有明確的藥理學(xué)作用, 而免疫原性較弱。Natpara是重組人甲狀旁腺激素, 在大鼠致癌性試驗中可見骨肉瘤發(fā)生率的增加, 且具有給藥劑量和給藥時間的相關(guān)性, 骨腫瘤的人體風(fēng)險不能排除; 因此, 在該藥品的說明書中, 不僅有關(guān)于骨腫瘤的黑框警告, 并且該藥的使用還納入了風(fēng)險評估和減低策略(Risk Evaluation and Mitigation Strategies, REMS), 以此來確保藥品獲益大于風(fēng)險[26]。瑞利珠單抗(Reslizumab)是一個針對IL-5的人源化單抗(IgG4 kappa), 即IL-5拮抗劑; 臨床前研究顯示, 該產(chǎn)品的藥理作用相關(guān)動物包括小鼠、兔和猴, 而親和力數(shù)據(jù)顯示, 人、猴、小鼠的KD值分別是24、20和31pM, 在小鼠重復(fù)給藥6個月的毒性試驗結(jié)果顯示, 隨著給藥時間的延長, 系統(tǒng)暴露量降低, 表明小鼠體內(nèi)有中和抗體產(chǎn)生。因此, 該產(chǎn)品最終是采用Tg.rasH2小鼠進行26周致癌性試驗替代野生型小鼠的2年致癌性試驗, 且豁免2年大鼠致癌性試驗[27]。
表 5 BLA申請中包含嚙齒動物致癌性試驗的情形

有趣的是, FDA在同一年批準了2個PCSK9 (Proprotein Convertase Subtilisin Kexin Type 9)抑制劑[28-29], 除了表 5所列的Amgen公司的Evolocumab, 另一個是S a n o f i-A v e n t i s公司的A l i r o c u m a b (BLA125559);然而, Evolocumab進行了2年地鼠致癌性試驗, 而Alirocumab未進行動物致癌性試驗。針對Alirocumab, 盡管采用大鼠進行致癌性試驗是可行的, 但申請人依據(jù)前期毒理研究結(jié)果和文獻報道申請豁免致癌性試驗:基于文獻, 可獲得PCSK9抑制劑與腫瘤不具有相關(guān)性, 降低膽固醇可通過改變腸道膽汁酸負荷或抑制免疫系統(tǒng)功能來影響腫瘤風(fēng)險的證據(jù)缺乏; 基于研究數(shù)據(jù), PCSK9抑制后與免疫抑制無關(guān)聯(lián)性, PCSK9抑制與腸道膽汁酸負荷之間無關(guān)聯(lián)性, 在毒理研究項目中均未見腫瘤或癌前病變。血漿膽固醇與大多數(shù)腫瘤風(fēng)險之間存在驚人的負相關(guān), 最有可能的解釋是所謂的臨床前腫瘤效應(yīng)。大量的證據(jù)支持這樣一種假設(shè), 即低膽固醇水平雖然與總體腫瘤風(fēng)險增加有關(guān), 但不太可能是這種風(fēng)險增加的原因。針對Evolocumab, 為解決相似的風(fēng)險擔(dān)憂, 進行了2年地鼠致癌性試驗, 每2周給藥1次, 劑量達到100 mg·kg-1, 皮下注射, 試驗期間可見總膽固醇和低密度脂蛋白膽固醇降低, 未見藥物相關(guān)腫瘤, 與臨床人體劑量相比(140 mg Q2W, 420 mg QM和420 mg Q2W), 暴露分別增加了38、15和6.6倍。
2.2.5 不要求但實際進行了致癌性試驗的情形
從進行致癌性試驗的69個藥物中, 可以看到一些藥物盡管進行了動物致癌性試驗, 但是從臨床應(yīng)用周期來看, 認為是不需要進行動物致癌性試驗的, 且這些致癌性試驗的結(jié)果在安全性評估中也沒有顯示出價值。具體包括:帕拉米韋(Peramivir, NDA206426)[30], 是一種流感病毒神經(jīng)氨酸酶抑制劑, 靜脈給藥用于治療2歲及以上出現(xiàn)癥狀不超過2天的急性非復(fù)雜流感, 進行了2年大鼠致癌性試驗, 而2年小鼠致癌性試驗因原研究單位(Johnson & Johnson)停止開發(fā)而試驗中止(未進行病理學(xué)檢查); 說明書中顯示"未進行靜脈注射給藥的致癌性研究, 進行了大鼠口服給藥2年的致癌性研究, 且未見藥物相關(guān)腫瘤發(fā)生"。Defibrotide (NDA208114)[31], 去纖苷酸, 是從豬的腸道組織中提取的一種以單鏈聚脫氧核糖核酸復(fù)合物為主的鈉鹽, 靜脈輸注給藥用于治療造血干細胞移植后發(fā)生的重度肝小靜脈閉塞癥, 最長給藥周期是60天; 但是, 在較早的年代(1989-1990年)進行了拌食給予大鼠和小鼠的2年致癌性試驗, 而在說明書中描述為"未進行靜脈給藥的致癌性研究"。Avatrombopag(NDA210238)[32], 促血小板生成素受體激動劑, 臨床口服給藥用于血小板減少癥的治療, 最長給藥周期是13天, 但是仍進行了2年大鼠和2年小鼠致癌性試驗, 盡管動物可見類胃癌發(fā)生率的增加, 但是認為類胃癌可能是由于長時間的高胃泌素血癥所致, 且高胃泌素相關(guān)的類胃癌在嚙齒動物中通常被認為是低風(fēng)險或與人類相關(guān)性較低。Moxidectin(NDA210867)[33], 一種驅(qū)蟲劑(盤尾絲蟲), 臨床上口服單次給藥, 非臨床試驗中仍進行了拌食給藥的2年大鼠和小鼠致癌性試驗, 因FDA統(tǒng)計部門未完成致癌性試驗數(shù)據(jù)的統(tǒng)計分析, 說明書中仍是"未確認長期致癌效應(yīng)"。Tafenoquine(NDA210795)[34], 是一種抗瘧藥, 用于間日瘧原蟲的根治(預(yù)防復(fù)發(fā)); 臨床單次用藥; 進行了2年小鼠和大鼠致癌性試驗, 說明書中顯示"由于臨床中單次用藥, 動物試驗的結(jié)果不能代表人體致癌性風(fēng)險"。Lofexidine (NDA209229)[35], 中樞α2腎上腺素能激動劑, 用于減輕阿片類藥物戒斷癥狀, 臨床最長給藥周期是14天, 同樣也是在較早的年代(1977-1978年)開展了2年大鼠和小鼠致癌性試驗, 但是均因未執(zhí)行GLP、未測定飼料中藥物含量以及高劑量不充分或動物死亡較多等原因認定2項試驗無效; 說明書中顯示"目前還沒有足夠的長期動物試驗來評估該產(chǎn)品的潛在致癌性"。
3 致癌性試驗類型的選擇
3.1 ICH S1B的要求
藥物致癌性試驗僅在獲得一定的關(guān)鍵信息后才進行, 包括遺傳毒性研究的結(jié)果、用藥人群、臨床用藥方案、動物和人體藥效動力學(xué)(選擇性、劑量-反應(yīng)關(guān)系)以及重復(fù)給藥毒性試驗結(jié)果。如果任何種屬(包括非嚙齒動物)的重復(fù)給藥毒性試驗可能表明受試物具有免疫抑制作用、激素活性或其他被認為對人體是一種危險因素的活性, 那么這類信息就應(yīng)在進一步評價潛在致癌性的試驗設(shè)計中予以考慮。
選擇具體致癌性試驗方法時應(yīng)具體問題具體分析。鑒于致癌過程的復(fù)雜性, 任何單一的試驗方法都無法預(yù)測所有人用藥物的潛在致癌性。基本方案包括一項長期嚙齒類動物致癌性試驗, 加上另一項附加的體內(nèi)致癌性試驗作為補充, 以提供長期致癌性試驗不易得到的其他信息; 除非是短期或長期致癌性試驗和遺傳毒性試驗以及其他數(shù)據(jù)提示一種藥物明確對人類具有致癌風(fēng)險, 通常不用再次進行致癌性試驗。而附加的體內(nèi)致癌性試驗包括:短期或中期嚙齒類動物體內(nèi)試驗系統(tǒng)(應(yīng)盡量使用能提供致癌終點的體內(nèi)模型, 包括嚙齒類啟動-促進模型、轉(zhuǎn)基因鼠致癌模型或新生嚙齒類動物致癌模型), 以及第二種屬嚙齒類動物長期致癌性試驗。
長期致癌性試驗的動物種屬選擇時, 應(yīng)考慮以下因素:藥理學(xué)、重復(fù)給藥毒性、受試物的代謝特性、毒代動力學(xué)和給藥途徑, 在缺乏確鑿證據(jù)時, 推薦選擇大鼠。
3.2 FDA批準的127項致癌性試驗分析
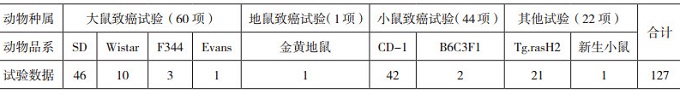
過去5年期間, FDA批準的213個NDA或BLA中, 共有69個新藥完成了127項動物致癌性試驗(詳見表 6)。與Wistar大鼠相比, 盡管SD大鼠存在2年生存率偏低的情況, 但在60項2年大鼠致癌性試驗中, 46/60項試驗仍是采用了SD大鼠; 這可能與該品系動物背景數(shù)據(jù)較為充足, 且重復(fù)給藥毒性試驗中多采用它有關(guān)。而同一個品系的SD大鼠又可以細分為Crl:CD(SD)和Hsd:SD等不同種群, 不同種群的大鼠在自發(fā)性腫瘤發(fā)生率甚至腫瘤類型上也存在輕微差別[36-37], 需要在動物選擇時給予關(guān)注。理想條件下應(yīng)采用與人類代謝特征盡可能相似的嚙齒類動物品系。在普通小鼠的2年致癌性試驗中, 更多的是采用封閉群CD-1小鼠, 較少采用雜交系小鼠。唯一的新生小鼠致癌性試驗, 是采用Swiss小鼠給藥1年的方式。在ICH S1B中推薦的轉(zhuǎn)基因模型包括p53+/-缺失模型、TgAC模型、TgHras2模型(或稱Tg.rasH2小鼠模型)和XPA缺失模型, 但是這些模型普遍有如下缺陷:(1)不能100%檢測出人類致癌劑, 無法明確區(qū)分鼠類致癌劑非人類致癌劑與人類致癌劑非鼠類致癌劑; (2)預(yù)測致癌劑的人類靶器官方面應(yīng)用有限, 如肝臟[38-39]; (3)對基因毒性和非基因毒性致癌物的反應(yīng)不一致(如TgAC和p53+/-缺失模型); (4)定量風(fēng)險預(yù)測方面的使用范圍有限[40]; (5)替代模型的遺傳背景對腫瘤的發(fā)生造成了影響[41], 如C57BL/6背景對肝癌有抑制作用, 導(dǎo)致該遺傳背景的模型對某些致癌劑不敏感。而每個模型小鼠均有其優(yōu)缺點; Tg.rasH2小鼠模型由于針對基因毒性和非基因毒性致癌劑均有較高的檢出率, 且對人類致癌劑非鼠類致癌劑較敏感[42-43], 在目前的新藥非臨床研究中, 較多采用的是"2年大鼠致癌性試驗+26周Tg.rasH2小鼠致癌性試驗"這種組合方式。需要提醒的是, 由于藥物種類不同, 藥理學(xué)特性及體內(nèi)代謝特點不同, 臨床應(yīng)用上的諸多差異, 在選擇具體的致癌性試驗類型時, 例如采用常規(guī)大、小鼠的2年生命周期給藥, 還是采用上述的"2年大鼠致癌性試驗+26周Tg.rasH2小鼠替代性致癌性試驗"的組合方式, 必須在致癌性試驗"特殊方案評估(Special Protocol Assessment, SPA)"過程中獲得FDA的認可或建議。關(guān)于FDA致癌性研究SPA的指導(dǎo)文件以及FDA內(nèi)部致癌性試驗評估執(zhí)行委員會(Executive Carcinogenicity Assessment Committee, E C A C)的功能和運作, 將在后續(xù)專題文章中詳述。
表 6 近5年FDA批準新藥的致癌性試驗類型信息
4 致癌性試驗的劑量選擇
4.1 ICH S1C(R2)的要求
關(guān)于高劑量的選擇, ICH S1C(R2)推薦的指標包括[3]:(1)基于毒性改變的指標:ICH安全性專家小組同意繼續(xù)使用最大耐受劑量(MTD)作為有用的致癌性試驗選擇高劑量的毒性指標, 該劑量是指致癌性試驗中預(yù)期產(chǎn)生可耐受的最小毒性作用的劑量, 包括與對照組相比體重的增長率減少不超過10%、有靶器官毒性、臨床病理參數(shù)的明顯改變等; (2)藥代動力學(xué)指標:測定血漿中游離藥物濃度是考察組織中游離藥物濃度最合適的間接測定方法, 而AUC是最全面的藥代動力學(xué)指標, 兼顧了化合物的血漿濃度和體內(nèi)滯留時間, 而基于以MTD進行的致癌性試驗數(shù)據(jù)庫的分析, 以嚙齒動物血漿中原型藥物和/或代謝產(chǎn)物AUC為人的25倍作為致癌性試驗的高劑量被認為是實用的; 特別注意的是要有藥物在嚙齒動物與人體中代謝相似的證據(jù), 以及在估計相對暴露量時, 應(yīng)考慮不同種屬之間蛋白結(jié)合率的差異; (3)吸收飽和量:可用藥物或其活性代謝物系統(tǒng)生物利用度計算得到的吸收飽和量作為致癌性試驗的高劑量, 同時低、中劑量也需考慮藥物代謝的飽和以及消除途徑; (4)藥效學(xué)指標:選擇的高劑量應(yīng)使動物產(chǎn)生的藥效學(xué)反應(yīng)足夠大, 以避免需要進一步遞增劑量, 然而, 該劑量不應(yīng)干擾生理或體內(nèi)平衡狀態(tài), 從而降低研究的價值, 如造成低血壓和抑制凝血等; (5)最大可行劑量:如果無法獲得MTD或無法達到25倍人體暴露量AUC時, 各個藥物監(jiān)管機構(gòu)均以最大可行劑量(MFD)作為可接受的指標。通過拌食法進行致癌性試驗時, 最大量是飼料量的5%;當(dāng)用其他途徑時, 還應(yīng)考慮到可行性和局部耐受性; (6)劑量限度:當(dāng)推薦人用最大劑量不超過500 mg·d-1時, 致癌性試驗的高劑量可設(shè)定在1500 mg·kg-1·d-1, 但是, 需要該劑量水平的系統(tǒng)暴露量比人體擬定治療劑量所能達到的暴露量至少高一個數(shù)量級(否則應(yīng)考慮增加暴露量或重新考慮動物模型); 如果人用劑量超過500 mg·d-1, 高劑量還可增加至MFD。
關(guān)于中、低劑量, 通常以倍率遞減方式來確定, 以期獲得劑量相關(guān)的致癌性反應(yīng)(如果藥物存在致癌性的話)。
4.2 2年大鼠致癌性試驗劑量選擇的實例分析
從FDA批準的54項2年大鼠致癌性試驗數(shù)據(jù)來看:絕大多數(shù)藥物(48/54)是依據(jù)MTD, 或AUC比值, 或二者的綜合分析來確定高劑量組給藥劑量; 4/54項試驗是依據(jù)MFD; 1/54項試驗是依據(jù)藥效學(xué)作用; 還有1/54項是采用六大標準之外的方法(局部藥物濃度)。劑量選擇的標準大多不是唯一性, 利用多種劑量選擇標準, 為設(shè)計最佳的藥物致癌性試驗提供了更大的靈活性。因篇幅有限, 本文僅將有代表性的幾個劑量選擇方式列舉如下:
Evolocumab(BLA125522), PCSK9單抗, 臨床給藥途徑是皮下注射, 人體最大推薦劑量(MRHD)是140 mg每兩周一次或420 mg每月一次。在地鼠重復(fù)皮下注射給藥13周的毒性試驗中, 給藥劑量分別是100 mg·kg-1和300 mg·kg-1; 結(jié)果顯示, 在100 mg·kg-1水平, 動物血清CHOL、HDL和LDL水平均達到最大抑制效應(yīng); 因此, 在2年致癌性試驗中, 采用皮下注射給藥, Q2W, 劑量分別是10、30和100 mg·kg-1; 結(jié)果顯示未見藥物相關(guān)腫瘤發(fā)生。
Plecanatide(SP-304, NDA208745)[44], 為含有16個氨基酸的化學(xué)合成多肽, 鳥苷酸環(huán)化酶C激動劑, 局部作用于腸上皮表面, 促進腸液分泌, 用于治療慢性特發(fā)性便秘, MRHD是3 mg·d-1。FDA的ECAC關(guān)于劑量設(shè)置的建議:基于300 mg·kg-1·d-1組動物有體重增長量降低, 推薦雌性動物劑量是10、30和100 mg·kg-1·d-1; 基于大鼠腸道局部藥物濃度較大, 達到了最大藥理學(xué)作用, 推薦雄性動物劑量是10、30和100 mg·kg-1·d-1。
巴瑞替尼(Baricitinib, NDA207924)[45], JAK1/2激酶抑制劑, 用于治療類風(fēng)濕關(guān)節(jié)炎, MRHD是2 mg·d-1。在大鼠重復(fù)灌胃給藥26周的毒性試驗中, 雄性劑量分別是0.5、5、25和100 mg· kg-1·d-1, 其中, 5和25 mg·kg-1·d-1組動物體重分別降低8%和16%;雌性劑量分別是0.5、5、25和100/60 mg·kg-1·d-1, 其中, 25 mg·kg-1·d-1組動物體重?zé)o降低, 而外周血淋巴細胞降低39%。根據(jù)以上信息, 2年大鼠致癌性試驗雄性動物劑量選擇1、3和8 mg·kg-1·d-1, 雌性動物劑量是3、8和25 mg·kg-1·d-1。
4.3 短期致癌性試驗的劑量選擇的實例分析
與2年大鼠致癌性試驗不同, 轉(zhuǎn)基因小鼠致癌性試驗更關(guān)注的是動物耐受性, 即基于毒性改變來選擇高劑量, 而非體內(nèi)暴露量[46-47]。因此, 在FDA最近5年期間批準的21項Tg.rasH2小鼠致癌性試驗中(詳見表 7), 有2項試驗的給藥劑量均大于或等于劑量限度法的1500 mg·kg-1·d-1, 分別是Dasabuvir(NDA206619)和Lumacaftor (NDA206038)。且與大鼠致癌性試驗結(jié)果的描述方式不同, 在藥品說明書中, 一般也不描述轉(zhuǎn)基因小鼠致癌性試驗中動物體內(nèi)暴露量與人體暴露量的比值或倍數(shù)。
表 7 過去5年期間FDA批準的21項Tg.rasH2小鼠致癌性試驗劑量設(shè)置信息

26周Tg.rasH2小鼠致癌性試驗的劑量選擇均依據(jù)Non-Tg.rasH2小鼠4周劑量探索試驗的結(jié)果, 同樣以Baricitinib為例[45], 在探索試驗中, 雄性和雌性動物給藥劑量均是75、150和300 mg·kg-1·d-1; 根據(jù)300 mg·kg-1劑量水平, 雄性動物淋巴細胞降低50%, 雌性動物出現(xiàn)的腎小管變性/壞死、擴張, 判定雄性和雌性動物的MTD分別為300和150 mg·kg-1·d-1。故在26周致癌性試驗中, 雄性動物給藥劑量分別是15、40和300 mg·kg-1·d-1, 雌性動物給藥劑量分別是10、30和150 mg·kg-1·d-1。結(jié)果顯示各組動物均未見藥物相關(guān)腫瘤發(fā)生。
5 結(jié)論
嚙齒類動物致癌性試驗的試驗周期長、費用高, 且致癌性試驗的復(fù)雜程度遠遠超出指導(dǎo)原則的要求, 藥品申請人需根據(jù)擬定的臨床用藥周期、藥物代謝特征、藥理毒理學(xué)研究結(jié)果等綜合考慮動物致癌性試驗的必要性; 結(jié)合臨床試驗進度和非臨床研究機構(gòu)的綜合能力合理選擇致癌性試驗的啟動時機。對于大分子蛋白等生物制品, 也不能想當(dāng)然地認為不需開展動物致癌性試驗, 更應(yīng)該是結(jié)合產(chǎn)品特點進行證據(jù)權(quán)重分析, 理性評估。當(dāng)前動物致癌性試驗的常規(guī)組合方式是2年大鼠致癌性試驗加上一項26周Tg.rasH2小鼠致癌性試驗, 但是, 兩項試驗在試驗設(shè)計、飼養(yǎng)條件、劑量選擇、統(tǒng)計分析等方面均存在區(qū)別, 研究者要結(jié)合實際案例加強指導(dǎo)原則學(xué)習(xí)。在進行致癌性試驗決策時, 除了ICH S1指導(dǎo)原則外, 還要兼顧ICH M3和ICH S6等指導(dǎo)原則中關(guān)于致癌性試驗的相關(guān)考慮。致癌性試驗不同于任何其他非臨床安全性研究, 藥物監(jiān)管機構(gòu)也會介入方案評估, 尤其是試驗類型、劑量設(shè)計等, FDA的ECAC會給予申請人最終的方案評估意見和建議, 我國也鼓勵申請人就致癌性試驗方案與國家藥品監(jiān)督管理局藥品審評中心進行溝通交流。因此, 申請人應(yīng)嚴格按照法規(guī)要求與監(jiān)管部門加強溝通交流, 取得監(jiān)管部門對方案評估的建議和指導(dǎo), 在實施ICH S1指導(dǎo)原則的過程中不斷積累和完善經(jīng)驗。另外, 目前ICH S1(R1)專家工作組正在針對致癌性試驗策略對S1指導(dǎo)原則進行修訂, 建議申請人和研究者密切關(guān)注ICH官網(wǎng)(www.ich.org/products/guidelines.html)公布的相關(guān)進展。
致謝
感謝軍事醫(yī)學(xué)科學(xué)院毒物藥物研究所的廖明陽教授對本文的指導(dǎo)和幫助!
參考文獻
[1] ICH. S1A Guidance for Industry: The Need for Long-term Rodent Carcinogenicity Studies of Pharmaceuticals[S]. 1996.
[2] ICH. S1B Guidance for Industry: Testing for Carcinogenicity of Pharmaceuticals[S]. 1997.
[3] ICH. S1C (R2) Guidance for Industry: Dose Selection for Carcinogenicity Studies of Pharmaceuticals[S]. 1997.
[4] FDA. Drug Approval Report[EB/OL].[2019-07-27]. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/.
[5] FDA. Pharmacology/Toxicology Review: Isavuconazonium[R]. NDA 207500. CDER, 2015.
[6] 于春榮, 笪紅遠, 王慶利. 直接抗丙肝病毒新藥的致癌性研究評價[J]. 中國新藥雜志, 2017, 26(1): 32-35.
[7] FDA. Pharmacology/Toxicology Review: Finafloxacin Otic Suspension[R]. NDA 206307. CDER, 2014.
[8] FDA. Pharmacology/Toxicology Review: Deoxycholic Acid[R]. NDA 206333. CDER, 2015.
[9] FDA. Pharmacology/Toxicology Review: Lifitegrast Ophthalmic Solution[R]. NDA 208073. CDER, 2016.
[10] FDA. Pharmacology/Toxicology Review: Vyzulta[R]. NDA 207795. CDER, 2017.
[11] FDA. Pharmacology/Toxicology Review: Rhopressa[R]. NDA 208254. CDER, 2017.
[12] FDA. Pharmacology/Toxicology Review: Patiromer[R]. NDA 205739. CDER, 2015.
[13] FDA. Pharmacology/Toxicology Review: Sodium Zirconium Cyclosilicate[R]. NDA 207078. CDER, 2018.
[14] FDA. Pharmacology/Toxicology Review: Cholbam[R]. NDA 205750. CDER, 2015.
[15] FDA. Pharmacology/Toxicology Review: Uridine Triacetate[R]. NDA 208169. CDER, 2015.
[16] FDA. Pharmacology/Toxicology Review: Insulin Degludec[R]. NDA 203314. CDER, 2015.
[17] FDA. Pharmacology/Toxicology Review: Aripiprazole Lauroxil[R]. NDA 207533. CDER, 2015.
[18] FDA. Pharmacology/Toxicology Review: Genvoya[R]. NDA 207561. CDER, 2015.
[19] FDA. Pharmacology/Toxicology Review: Emflaza[R]. NDA 208684. CDER, 2017.
[20] FDA. Pharmacology/Toxicology Review: Deutetrabenazine[R]. NDA 208082. CDER, 2017.
[21] ICH. M3(R2) Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals[S]. 2009.
[22] ICH. S6(R1) Preclinical Safety Evaluation of Biotechnologyderived Pharmaceuticals[S]. 2011.
[23] FDA. Pharmacology/Toxicology Review: Ixekizumab[R]. BLA125521. CDER, 2016.
[24] FDA. Pharmacology/Toxicology Review: Dulaglutide[R]. BLA125469. CDER, 2014.
[25] FDA. Pharmacology/Toxicology Review: Semaglutide[R]. BLA125511. CDER, 2015.
[26] FDA. Pharmacology/Toxicology Review: Natpara[R]. BLA209637. CDER, 2017.
[27] FDA. Pharmacology/Toxicology Review: Reslizumab[R]. BLA761033. CDER, 2016.
[28] FDA. Pharmacology/Toxicology Review: Evolocumab[R]. BLA125522. CDER, 2015.
[29] FDA. Pharmacology/Toxicology Review: Alirocumab[R]. BLA125559. CDER, 2015.
[30] FDA. Pharmacology/Toxicology Review: Peramivir[R]. NDA 206426. CDER, 2014.
[31] FDA. Pharmacology/Toxicology Review: Defibrotide[R]. NDA 208114. CDER, 2016.
[32] FDA. Pharmacology/Toxicology Review: Avatrombopag[R]. NDA 210238. CDER, 2018.
[33] FDA. Pharmacology/Toxicology Review: Moxidectin[R]. NDA 210867. CDER, 2018.
[34] FDA. Pharmacology/Toxicology Review: Tafenoquine[R]. NDA 210795. CDER, 2018.
[35] FDA. Pharmacology/Toxicology Review: Lofexidine[R]. NDA 209229. CDER, 2018.
[36] 張素才, 芮志佩, 張冬霞, 等. 長期致癌試驗動物飼養(yǎng)管理技術(shù)關(guān)注要點[J]. 中國新藥雜志, 2017, 26(2): 162-168.
[37] Brower M, Grace M, Kotz C, et al. Comparative Analysis of Growth Characteristics of Sprague Dawley Rats Obtained from Different Sources[J]. Lab Anim Res, 2015, 31(4): 166-173. DOI:10.5625/lar.2015.31.4.166
[38] Gulezian D, Jacobson-Kram D, Mccullough C, et al. Use of Transgenic Animals for Carcinogenicity Testing:Considerations and Implication for Risk Assessment[J]. Toxicol Pathol, 2000, 28: 482-499. DOI:10.1177/019262330002800320
[39] D Vries A, Van Oostrom C, Dortant P, et al. Spontaneous Liver Tumors and Benzo[a] Pyrene-induced Lymphomas in XPA-deficient Mice[J]. Mol Carcinog, 1997, 19: 46-53. DOI:10.1002/(SICI)1098-2744(199705)19:1<46::AID-MC7>3.0.CO;2-L
[40] Boverhof D, Chamberlain M, Elcombe C, et al. Transgenic Animal Models in Toxicology Historical Perspectives and Future Outlook[J]. Toxicol Sci, 2011, 121: 207-233. DOI:10.1093/toxsci/kfr075
[41] Storer R, Sistare F, Reddy M, et al. An Industry Perspective on the Utility of Short-term Carcinogenicity Testing in Transgenic Mice in Pharmaceutical Development[J]. Toxicol Pathol, 2009, 38: 51-61.
[42] Paranjpe M, Belich J, Mann P, et al. A Comparison of Spontaneous Tumors in Tg.rasH2 Mice in 26-week Carcinogenicity Studies Conducted at a Single Test Facility During 2004 to 2012 and 2013 to 2018[J]. Toxicol Pathol, 2019, 47(1): 18-25. DOI:10.1177/0192623318810202
[43] 宋征, 徐景宏, 王慶利, 等. 轉(zhuǎn)基因小鼠在藥物致癌性評價中的應(yīng)用[J]. 中國藥理學(xué)與毒理學(xué)雜志, 2010, 24(6): 557-561.
[44] FDA. Pharmacology/Toxicology Review: Plecanatide[R]. NDA 208745. CDER, 2017.
[45] FDA. Pharmacology/Toxicology Review: Baricitinib[R]. NDA 207924. CDER, 2018.
[46] Prashant R, Namblar, and Daniel M, et al. Regulatory Forum Commentary Counterpoint:Dose Selection for RasH2 Mouse Carcinogenicity Studies[J]. Toxicol Pathol, 2015, 43(5): 628-632. DOI:10.1177/0192623315578012
[47] Jarig Darbes, Frank D Sistare, Joseph J D. Regulatory Forum Commentary Counterpoint:Dose Selection for RasH2 Mouse Carcinogenicity Studies[J]. Toxicol Pathol, 2015, 43(5): 621-627. DOI:10.1177/0192623315587722

來源:xml-data


