您當前的位置:檢測資訊 > 科研開發(fā)
嘉峪檢測網(wǎng) 2019-04-22 09:57
IVDR將于2022年5月26日強制執(zhí)行,很多體外診斷試劑廠商覺得時間還早,法規(guī)狗卻深深覺得已經(jīng)不早了,因為相比較MDR對于醫(yī)療器械企業(yè)的影響,IVDR對于體外診斷試劑的廠商影響更大。為什么呢?
IVDD時代只有20%左右的體外診斷試劑是需要公告機構(gòu)發(fā)證的,還有80%企業(yè)可以自我聲明。到了IVDR這兩個比例會倒過來,也就是80%左右的體外診斷試劑需要公告機構(gòu)發(fā)證,20%左右企業(yè)自我聲明。
舉個例子,腫瘤標志物CA19-9檢測的試劑盒,在IVDD法規(guī)下是自我聲明,到了IVDR就需要公告機構(gòu)發(fā)證。既然影響這么大,體外診斷試劑的廠商又該如何應(yīng)對呢?法規(guī)狗認為應(yīng)從以下8個問題入手,分別是分類,標準,科學有效性,分析性能,臨床性能,上市后性能跟蹤,UDI和公告機構(gòu)。

IVDD將體外診斷醫(yī)療器械分為三類:List A、List B、自我檢測(Self-test)和其它(others)。
其中,List A包含:1)血型分型試劑(用于確定ABO系統(tǒng)和rhesus(C,c,D,E,e)anti-Kell系統(tǒng));2)艾滋病(含HIV1/2)、人體T細胞白血病病毒(含HTLV I/II)、乙型肝炎、丙型肝炎、丁型肝炎的診斷試劑。
List B包含:1)血型分型試劑((anti-Duffy和anti-Kidd系統(tǒng));2)抗紅細胞不規(guī)則抗體檢測試劑;3)傳染病類(風疹、弓形蟲、巨細胞病毒、衣原體)診斷試劑;4)腫瘤標志物類(前列腺特異抗原)診斷試劑;5)21三體綜合征檢測試劑、苯丙酮尿癥診斷試劑;6)HLA組織類別(DR/A/B)檢測試劑;7)自我診斷血糖檢測試劑。
IVDR參照GHTF的分類思路按照風險等級將IVD產(chǎn)品分為4類:Class A(風險最低)、Class B、Class C和Class D(風險最高),涉及血型分型、血液或組織相容性、傳染性疾病、腫瘤篩查、伴隨診斷、基因檢測、先天性疾病篩查、自我檢測、IVD分析儀器等細分領(lǐng)域;并根據(jù)產(chǎn)品的預期用途和被測量的分析物制定了7條分類規(guī)則,相比于舊版IVDD,大大擴充了產(chǎn)品的分類范圍。如果產(chǎn)品符合多條分類規(guī)則,則按照“就高不就低”的原則進行分類。
IVDD法規(guī)規(guī)定List A試劑,List B試劑及自我測試的試劑需要由公告機構(gòu)發(fā)證。IVDR法規(guī)規(guī)定B,C,D類的體外診斷試劑都需要由公告機構(gòu)發(fā)證。這就回應(yīng)了引子里說的80/20的事情。
所以體外診斷試劑的廠商應(yīng)先搞清楚自己的產(chǎn)品在IVDR的分類,如果不是A,就需要和公告機構(gòu)盡早取得聯(lián)系了。
在做體外診斷試劑的CE認證時我們首先要意識到歐盟和國內(nèi)標準的差異,我們都知道歐盟有自己的協(xié)調(diào)標準,如果你抱著GB和YY可能是不夠的。舉個例子:
法規(guī)狗有看過一些國內(nèi)的體外診斷試劑的技術(shù)審查指導原則,舉個例子來說《C-肽測定試劑注冊技術(shù)審查指導原則》中有提到空白限,在該原則中有提到空白限是這樣做的:
空白限的確定常使用同批號試劑對零濃度校準品(或校準品稀釋液)進行至少20 次重復檢測,計算所得信號值均值和標準差(SD),將(X +2SD)帶入劑量—反應(yīng)曲線,計算出的濃度 值即為空白限。
這個空白限法規(guī)狗理解為是檢測限。如果C-肽測定試劑盒要去做CE認證,你會發(fā)現(xiàn)CE對于檢測限的要求更高,檢測限可以衍生出三個:
LOB: Limit of blank, 這個就是上述原則中提到的空白限。
LOD: Limit of detection, measured quantity value, obtained by a given measurement procedure, for which the probability of falsely claiming the absence of a measurand in a material is β, given a probability α of falsely claiming its presense.( 通過給定的測量程序獲得的測量量值,在該測量值下,錯誤地宣稱不含分析物(實際上有分析物)的概率為β,錯誤地宣稱含分析物(實際上不含分析物)的概率為α) 。
LOQ: Limit of quantitation,Lowest amount of a measurand in a material that can be quantitatively determined with stated accuracy. (材料中測量值的最低值,可以用所宣稱的準確度定量確定)。
我相信看到這里大家一定是頭大了,就是一個檢測限扯出這么多問題。如果你按照cFDA的思路去做只有LOB看上去是符合要求了,但你再細看呢?它就做了20次重復,那為什么20次重復就夠了呢?這些問題你在做CE認證的時候要基于科學依據(jù)去回答,而不是僅僅告訴審核員我就做了20次重復,要告訴它為什么,并且你的理由是站得住腳的。
所以廠商要基于自己的產(chǎn)品去查詢有哪些協(xié)調(diào)標準是要滿足的。在這里法規(guī)狗提前通告一下,了解了你的產(chǎn)品要滿足的協(xié)調(diào)標準之后你發(fā)現(xiàn)這些協(xié)調(diào)標準還會指向CLSI的標準,因此你還要把CLSI的標準找出來。
CLSI是美國【臨床實驗室標準化協(xié)會】的英文縮寫,英文名為Clinical and Laboratory Standards Institute。CLSI前身是NCCLS【美國臨床實驗室標準化委員會】,英文名稱為National committee for clinical laboratory。
美國CLSI的抗微生物藥物敏感性試驗操作方法和判斷標準,是國內(nèi)臨床細菌檢驗遵循的標準。由于制訂該項標準需要投入大量的人力、財力和物力,所以大多數(shù)國家包括中國都還沒有能力建立自己的標準而依賴CLSI的方法和標準。CLSI標準經(jīng)常更新,是否讓你想起了CE法規(guī)的State of the art的精神。

科學有效性( Scientific validity)是在IVDR中出現(xiàn)的新詞匯,到底是什么意思呢? 法規(guī)狗查詢了一下維基百科,定義是這樣的: Scientific validity is the applicability of a conclusion drawn in the context of a scientific experiment to the world at large. 翻譯一下: 科學有效性是指科學實驗在全世界范圍內(nèi)得出的結(jié)論的適用性。
通俗一點說,廠商需要找大量的信息來證明試劑的檢測方法是科學合理的。那么要如何來說明這個問題呢? 法規(guī)狗認為主要是要說明樣本中的分析物和某種疾病或者某種疾病的發(fā)展狀態(tài)等等之間的關(guān)系是科學有效的。
舉個例子,對于糖化血紅蛋白HbA1c檢測的試劑盒而言, 你要證明糖化血紅蛋白H1bA1c和糖尿病之間的關(guān)系,換句話說通過檢測H1bA1c的含量來得知病人是否有潛在的得糖尿病的可能性是科學有效的。你可能會說這不是顯而易見的嘛,那就請你提供證據(jù)。
證據(jù)可以從哪里來呢? 在IVDR的法規(guī)中也提到了證據(jù)主要來自于以下幾個方面:
針對于同一分析物或者標志物的科學合理性的相關(guān)信息(你就理解為如果你之前準備了這個資料,下一次類似產(chǎn)品要準備可以參考之前的資料)
科學文獻
專家意見
研究結(jié)果(Results from proof of concept studies)
臨床性能研究結(jié)果
可以看出后續(xù)為了證明科學有效性,至少要查詢大量的文獻。基于法規(guī)狗的經(jīng)驗,查文獻絕對是一門技術(shù)活。
IVDR附錄I的9.1(a)有提到分析性能的要求,主要要考慮以下分析性能指標:
測量的準確度( Accuracy of measurement)
測量的正確度(Trueness of measurement)
測量的精密度(Precision of measurement)
法規(guī)狗經(jīng)常用打靶的例子來解釋準確度和精密度,請見下圖:
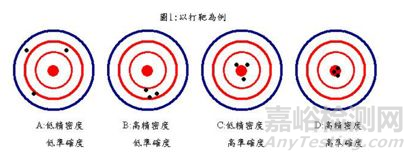
正確度和準確度是不同的,不同在哪里呢? 我們來看定義:
Accuracy(準確度): Closeness of agreement between a test result and the accepted reference value. (EP17-A)
Trueness(正確度): The closeness of agreement between the average value obtained from a large series of test results and an accepted reference value; NOTE: The measure of trueness is usually expressed in terms of bias.
可以看出準確度關(guān)注的是單次測量值,而正確度關(guān)注的是平均測量值。正確度會影響準確度。
分析敏感性(Analytical sensitivity)就是我們平常講的檢測限,但是CE的檢測限的含義和國內(nèi)有較大不同
分析特異性(Analytical specificity)主要要評估干擾物質(zhì)和交叉反應(yīng)物質(zhì)對于檢測結(jié)果的影響,干擾物質(zhì)分為內(nèi)源性的和外源性的,內(nèi)源性的包括: 血紅蛋白,脂類,膽紅素等等。外源性只要指你吃進去喝進去的東西,比如: 藥品,酒精,食物等等。交叉反應(yīng)物質(zhì)主要是指: 和待測分析物類似的蛋白,或者待測分析物的代謝產(chǎn)物等等。
線性范圍及可測量范圍
臨界值(Cut-off value)
有兩點要提起注意: 1,在進行分析性能評估之前先要做方案;2, 定性體外診斷試劑和定量體外診斷試劑分析性能評估的內(nèi)容會有很大不同。
IVDR附錄I的9.1(b)有提到體外診斷試劑主要要考慮以下臨床性能,主要包括:
敏感性(Sensitivity): 定性體外診斷試劑檢測結(jié)果是陽性的數(shù)量占金標準檢測結(jié)果是陽性數(shù)量的百分比。
特異性(Specificity): 定性體外診斷試劑檢測結(jié)果是陰性的數(shù)量占金標準檢測結(jié)果是陰性數(shù)量的百分比。
陽性預測值(Positive predictive value, PPV)是指篩檢試驗檢出的全部陽性例數(shù)中,真正“有病”的例數(shù)(真陽性)所占的比例,反映篩檢試驗結(jié)果陽性者患目標疾病的可能性。
陰性預測值(negative predictive value, NPV)指檢驗結(jié)果為陰性的受試者中真正未患病的比例。診斷試驗的預測值受到敏感度、特異度和受試者中患病率的影響。
陽性似然比(Positive likelihood ratio)是篩檢結(jié)果的真陽性率與假陽性率之比。說明篩檢試驗正確判斷陽性的可能性是錯誤判斷陽性可能性的倍數(shù)。比值越大,試驗結(jié)果陽性時為真陽性的概率越大。
陰性似然比(Negative likelihood ratio)是篩檢結(jié)果的假陰性率與真陰性率之比。表示錯誤判斷陰性的可能性是正確判斷陰性可能性的倍數(shù)。其比值越小,試驗結(jié)果陰性時為真陰性的可能性越大。
上述六個指標基本上是針對于定性的體外診斷試劑的,我們用一個例子來和各位解釋一下。
|
金標準 |
總數(shù) |
|||
|
陽性 |
陰性 |
|||
|
待評估試劑 |
陽性 |
真陽性(TP) |
假陽性(FP) |
TP+FP |
|
陰性 |
假陰性(FN) |
真陰性(TN) |
FN+TN |
|
|
總數(shù) |
TP+FN |
FP+TN |
N |
|
預計敏感性(Sens)= 100X [TP/(TP+FN)]
預計特異性(Spec)= 100X[TN/(FP+TN)]
陽性預測值= 100X[TP/(TP+FP)]
陰性預測值= 100X[TN/(FN+TN)]
陽性似然比=Sens/(1-Spec)
陰性似然比=(1-Sens)/Spec
還有一個指標是和定量的體外診斷試劑相關(guān)的:正常人群和得病人群的某一指標的預計值
上述這些數(shù)據(jù)很大程度上是需要通過臨床試驗獲得的,有一些醫(yī)療器械廠商有可能在國內(nèi)做了臨床,那么你要考慮國內(nèi)的臨床試驗數(shù)據(jù)是否可以用于CE的申請,因為畢竟可能會有人種和流行病學的差異
如果接觸過上市后臨床跟蹤的廠商就會很容易理解體外診斷試劑的上市后性能跟蹤(PMPF, post-market performance follow-up)。思路和上市后臨床跟蹤是一樣的,我不知道產(chǎn)品上市后大規(guī)模使用會出現(xiàn)什么問題,因此法規(guī)要求體外診斷試劑的廠商持續(xù)關(guān)注產(chǎn)品上市后的臨床表現(xiàn)。那么這個工作主要可以通過哪些方式去做呢?
IVDR的附錄XIII的Part B給出了具體的信息:
收集客戶的信息
收集文獻的信息
同行實驗室對比試驗(Ring trial)
流行病學研究
檢索臨床試驗數(shù)據(jù)庫
開展上市后臨床試驗
等等
如果你認為你的試劑不需要做PMPF那么請給出充足的理由。關(guān)于PMPF法規(guī)狗預計后續(xù)歐盟會出臺指南文件,因為目前給出的信息沒有太多的實際指導意義。
對于UDI(Unique device identifier)我相信大家或多或少都有一些了解。法規(guī)狗在很多場合講過UDI就是醫(yī)療器械的“身份證”,這個“身份證”可以方便醫(yī)療器械的追溯。MDR中對于UDI有明確的要求。
滿足UDI,體外診斷試劑的廠商需要考慮兩個方面的要求,一個方面是內(nèi)部要建立一套UDI的編碼系統(tǒng),另外一個方面醫(yī)療器械廠商需要把在歐盟銷售的產(chǎn)品的UDI信息在歐盟的數(shù)據(jù)庫中做登記。
第一方面該如何在內(nèi)部建立一套UDI的編碼系統(tǒng)呢?廠商首先要確定選用哪一家的編碼系統(tǒng),在MDR的條款120中已經(jīng)提到除非歐盟再特別指定一些UDI的發(fā)證機構(gòu),那么目前比較成熟的三家發(fā)證機構(gòu)GS1, HIBCC和ICCBBA就被認為是歐盟認可的UDI發(fā)證機構(gòu)。
國內(nèi)的醫(yī)療器械廠商對GS1應(yīng)該不陌生。GS1就是國際物品編碼協(xié)會,該協(xié)會的主要目的是促進客戶、合作伙伴和供應(yīng)商之間以一套供應(yīng)鏈標準語言進行溝通,GS1有主導很多供應(yīng)鏈標準的制定。我國的物品編碼中心代表中國加入了GS1, 負責推廣國際通用的、開放的、跨行業(yè)的全球統(tǒng)一編碼標識系統(tǒng)和供應(yīng)鏈管理標準,向社會提供公共服務(wù)平臺和標準化解決方案。因此如果國內(nèi)的醫(yī)療器械廠商想采用GS1的系統(tǒng),就可以和中國物品編碼中心聯(lián)系。
另一方面廠商還需要把產(chǎn)品的UDI信息在歐盟的數(shù)據(jù)庫中做登記,這個應(yīng)該不是很難。在MDR的附錄VI的B部分已經(jīng)非常清楚地列出廠商需要提交哪些信息,看上去等到數(shù)據(jù)庫正式運行之后直接去做遞交就可以了。
有一點法規(guī)狗要提醒大家,因為UDI涉及到所有的產(chǎn)品,因此法規(guī)狗推測提交UDI的信息的截止時間會因產(chǎn)品分類不同而不同,F(xiàn)DA執(zhí)行UDI的要求計劃要花7年左右的時間(2013-2020),估計歐盟要花的時間也不會少。因此醫(yī)療器械廠商要密切關(guān)注和UDI相關(guān)法規(guī)的出臺。
截止04/19/2019有IVDD發(fā)證資質(zhì)的公告機構(gòu)總共只有22家,你是不是還覺得挺多,這22家里面除掉英國的公告機構(gòu)(因為脫歐),再除掉沒有在中國設(shè)辦公室的,你會發(fā)現(xiàn)你能選擇的只有兩家(請見下圖)。
因此對于廣大中國體外診斷試劑廠商而言,預計會遇到粥多僧少的局面,法規(guī)狗覺得現(xiàn)在是那些還未開辟中國市場的公告機構(gòu)的絕佳機會。對于那些真正要往歐洲銷售的廠商來說,及時關(guān)注公告機構(gòu)拿資格的動態(tài)信息,并及早做準備尤為重要。



洋洋灑灑講了這么多,我再把重點提煉一下,趕緊把你的體外診斷試劑的分類搞清楚,如果需要公告機構(gòu)介入(粗略地說80%的可能性需要), 那么要盡早準備。你們是比醫(yī)療器械廠商多兩年,但是你們之前落下了太多的功課。人在江湖走,遲早都是要還的。

來源:啟升資訊


