您當前的位置:檢測資訊 > 法規(guī)標準
嘉峪檢測網(wǎng) 2024-11-14 08:33
一、基本概況
1、簡要介紹
歐盟由27個成員國組成,面積414萬平方公里,范圍覆蓋歐洲大部分地區(qū),西起大西洋、東臨獨聯(lián)體、南到地中海、北跨波羅的海,囊括除英國、獨聯(lián)體、西巴爾干地區(qū)外幾乎所有歐洲國家。歐盟總部設在比利時首都布魯塞爾。
2、人口和行政區(qū)劃
1、人口分布
歐盟27個成員國人口總計約4.5億,人口數(shù)量前五位的國家依次為:德國、法國、意大利、西班牙、波蘭。
2、成員國
目前,歐盟27個成員國包括奧地利、比利時、保加利亞、塞浦路斯、捷克、克羅地亞、丹麥、愛沙尼亞、芬蘭、法國、德國、希臘、匈牙利、愛爾蘭、意大利、拉脫維亞、羅馬尼亞、立陶宛、盧森堡、馬耳他、荷蘭、波蘭、葡萄牙、斯洛伐克、斯洛文尼亞、西班牙、瑞典。
3、2024年出口概況
2024年1-6月,中國向歐盟國家出口醫(yī)療器械總計約405.80億人民幣,同比增長約10.11%。
數(shù)據(jù)來源:廣州眾成大數(shù)據(jù)科技有限公司
2024年1-6月,歐盟EUDAMED新增收錄醫(yī)療器械產(chǎn)品總計47956款,其中4624款由中國企業(yè)自主申報。
數(shù)據(jù)來源:普瑞純證醫(yī)療科技(廣州)有限公司
二、歐盟醫(yī)療器械監(jiān)管機構和法規(guī)要求
歐盟主管當局及公告機構共同負責歐盟醫(yī)療器械的監(jiān)督管理。其中,公告機構負責對醫(yī)療器械上市的認證;主管當局負責公告機構資質(zhì)的認可。產(chǎn)品上市后的監(jiān)管由主管當局及公告機構共同負責執(zhí)行。
歐盟醫(yī)療器械認證需要遵循醫(yī)療器械法規(guī)(MDR, REGULATION (EU) 2017/745)要求。
目前已獲得有效醫(yī)療器械法規(guī)(MDR, REGULATION (EU) 2017/745)發(fā)證資質(zhì)的公告機構共50家,可通過以下歐盟官方網(wǎng)址查詢公告機構及其資質(zhì)范圍信息。
歐盟醫(yī)療器械法規(guī)(MDR, REGULATION (EU) 2017/745)于2017年5月發(fā)布,并在2021年5月強制執(zhí)行。同時截至目前為止,歐盟MDCG小組已發(fā)布近140份對法規(guī)解讀的相關MDCG指南和Q&A,為各相關方(制造商、歐代、公告機構、進口商)提供了深入的法規(guī)解讀和實用的指導,有助于各相關方更好地理解和執(zhí)行、并滿足歐盟醫(yī)療器械法規(guī)(MDR, REGULATION (EU) 2017/745)的要求。MDCG 指南和Q&A查詢網(wǎng)址。
三、醫(yī)療器械定義
歐盟醫(yī)療器械法規(guī)(MDR, REGULATION (EU) 2017/745)明確醫(yī)療器械(Medical device )定義如下:
◆ 用于人類儀器、器具、設備、軟件、植入物、試劑材料其他物品,其預期使用由制造商確定,不論單獨使用或組合使用,以達到下列一個或多個特定的醫(yī)療目的:
- 疾病的診斷、預防、監(jiān)測、預測、預后、治療或緩解。
- 損傷或殘疾的診斷、監(jiān)測、治療、緩解或補償。
- 對解剖學或生理或病理過程或狀態(tài)的查驗、替代或改變。
- 通過對取自人體(包括器官、血液和組織捐獻)的樣本進行體外檢查的方式來提供信息,而且不是通過藥理學、免疫學或新陳代謝的方式在人體內(nèi)或人體上實現(xiàn)其主要預期作用,但可以通過這些方式輔助其功能。
◆ 下列產(chǎn)品也應視為醫(yī)療器械:
- 控制或支持妊娠的器械;
- 專門用于第1(4)條所述器械和本點第一段所述器械的清潔、消毒或滅菌的產(chǎn)品。
相對較于原歐盟MDD指令,現(xiàn)MDR(REGULATION (EU) 2017/745)擴大了醫(yī)療器械范圍,例如明確了進行器械清潔、消毒、滅菌的產(chǎn)品也屬于醫(yī)療器械范圍,增加無預期醫(yī)療用途產(chǎn)品,如隱形眼鏡、用于減少/去除或破壞脂肪組織的器械、用于換膚、紋身或脫毛或其他皮膚治療的激光和強脈沖光器械等,具體內(nèi)容可參考MDR法規(guī)附錄XVI清單;此外,還包括專門用于替代器械部件或組件和顯著改變器械性能或安全特征或預期用途的器械(Article 23(2) MDR)。
四、醫(yī)療器械分類
1、依據(jù)
依據(jù)(REGULATION (EU) 2017/745)附錄VIII對醫(yī)療器械進行分類,共有22條分類規(guī)則:Rule 1 - Rule 4 非侵入性器械,Rule 5 - Rule 8 侵入性器械,Rule 9 - Rule 13有源器械,Rule 14 - Rule 22 特殊規(guī)則。
同時,制造商在進行醫(yī)療器械分類時可參考MDCG小組發(fā)布的如下分類界定指南文件進行:
◆ MDCG 2021-24 Guidance on classification of medical devices - 對MDR Article 2及MDR附錄VIII的補充性解釋
◆ MDCG 2022-5 Guidance on borderline between medical devices and medicinal products under Regulation (EU) 2017/745 on medical devices
◆ Background note on the use of the Manual on borderline and classification for medical devices under the Directives
◆ Manual on borderline and classification under Regulations (EU) 2017/745 and 2017/746 v2 - 分類界定結(jié)果(每3個月更新一次)
◆ MDCG 2019-11 Guidance on Qualification and Classification of software in MDR and IVDR
此外,如對產(chǎn)品分類有議時可聯(lián)系歐盟主管當局進行商討;具體無法確定分類的器械申請分類界定的工作流程可參考“Helsinki Procedure for borderline and classification under MDR & IVDR”。
2、分類
根據(jù)產(chǎn)品的風險等級,歐盟MDR (REGULATION (EU) 2017/745)仍將醫(yī)療器械分為四類,按風險由低到高依次為:I 類, IIa類,IIb 類,III類,I類器械又分為普通I 類及Is(無菌的I類器械),Im(有測量功能的醫(yī)療器械)及Irs(可重復使用手術器械)。
相比于原MDD指令,MDR增加了I類可重復使用手術器械(Irs)類別。其中,普通I 類器械可由制造商自我聲明,其它分類的器械均必須要公告機構的強制性認證。
五、認證流程
1、注冊流程解讀/流程圖

目前歐盟公告機構的認證流程基本一致:包括技術文檔審核(含臨床審核)、體系現(xiàn)場審核(分一階段和二階段審核)、公告機構內(nèi)部發(fā)證評審等幾個階段。
注:
1. TD(Techincal document 技術文檔)
2. 各公告機構的報價在技術文檔評審次數(shù)的涵蓋范圍上存在差異。部分公告機構的報價涵蓋了所有評審次數(shù),而另一些公告機構的報價僅包含一次評審的費用,若后續(xù)不合格整改后再次評審,則需另行繳費。
2、認證周期
關于MDR認證周期,歐盟至今沒有一個統(tǒng)一的標準流程、標準時間或統(tǒng)一的官方承諾,這是因為MDR認證過程涉及多個方面,包括產(chǎn)品的風險等級(如IIa,IIb,IIb植入,III類)各個公告機構的發(fā)證周期及服務承諾不同(這也是市場競爭的要素之一),以及后續(xù)制造商可能的整改周期等。
一般來講,大多數(shù)市場上反饋的認證周期約為1年半至兩年(成熟技術的IIa/IIb類產(chǎn)品)。然而,就當前實際狀況而言,歐盟當下的醫(yī)療器械法規(guī)(MDR)認證資源相對緊缺,故而建議制造商盡早開啟MDR認證流程,以便早日獲取證書。
3、認證費用
歐盟目前沒有一個統(tǒng)一的醫(yī)療器械認證費用標準。由于公告機構之間的市場競爭以及各自不同的服務模式,不同的公告機構認證費用存在差異。此外,公告機構在估算認證費用時,還需綜合考慮多個因素,如產(chǎn)品的風險等級(如Ⅱa,Ⅱb), 技術文檔的劃分,產(chǎn)品的技術特性(如是否包含WIFI功能、是否要求無菌處理)以及制造商的生產(chǎn)規(guī)模等。
一般來說,一個普通的Ⅱa 類產(chǎn)品的認證費用(一套技術文檔)會在30w-45w之間。具體情況需與公告機構溝通。
前四大公告機構收費情況可在附件點擊鏈接了解詳情。
◆ NB2797 BSI (NL)
◆ NB0123 TUV SUD (DE)
◆ NB0197 TUV Rheinland (DE)
◆ NB1639 SGS (BE)
4、符合性評估路徑
醫(yī)療器械的上市路徑會根據(jù)其分類的不同而有所差異。MDR(Regulation (EU) 2017/745)規(guī)定了不同分類產(chǎn)品所需的上市前審查和認證流程。
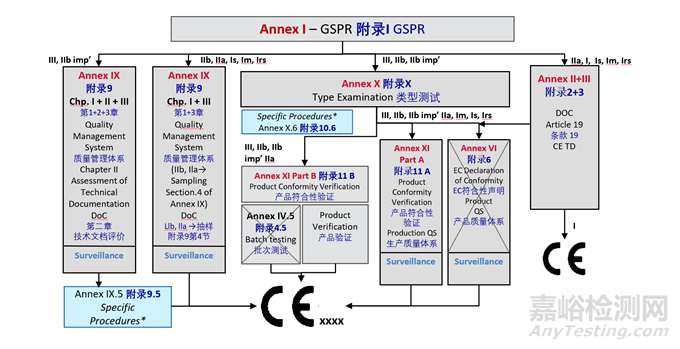

5、認證提交的技術文檔
根據(jù)MDR 2017/745 Annex II要求,制造商須建立一套精心組織、清晰明了、避免誤解、易于檢索的產(chǎn)品技術文檔(Technical document)。
1、上市前技術文檔(MDR附錄II要求)
a) 產(chǎn)品描述及規(guī)格(包括設備描述,已有設備和市場上類似設備,Basic UDI-DI和 EMDN code,技術文件所覆蓋的器械設備分類 Annex VI,器械歷史以及類似產(chǎn)品是否在歐盟或其它市場上市)
b) 制造商提供的信息(如產(chǎn)品標簽,說明書)
c) 設計及制造資料
d) 基本安全和性能要求GSPR
e) 收益風險分析和風險管理
f) 器械驗證和確認(臨床前測試數(shù)據(jù)及臨床數(shù)據(jù))
g) 符合性聲明DoC
2、上市后技術文檔(MDR 附錄II要求)還需依據(jù)上市后的信息積極更新內(nèi)容:
a) 上市后監(jiān)管計劃(條款84)
b) 定期安全性更新報告PSUR(條款 86)
c) 上市后監(jiān)督報告(條款85)
d) 風險管理(條款83)
e) 臨床評價(條款83)
f) 安全和和臨床性能總結(jié)(條款32、83,class III和植入器械(非定制/研究器械))
g) 上市后臨床跟蹤(附錄XIV, Part B)
h) 檢測并報告趨勢(條款88)
3、制造商義務
制造商應保存技術文檔、歐盟符合性聲明、適用時還有相關證書及修訂件和補充件的副本,在歐盟符合性聲明中所涵蓋的最后器械上市后,該文檔應至少向主管機構開放10年。若為植入器械,周期應至少為最后器械已投放市場后的15年。
若主管當局要求,制造商應提供完整的技術文件或其概要。
在歐盟境外注冊的制造商應指定歐盟境內(nèi)唯一的授權代表,并確保其有永久可用的技術文檔。
4、技術文檔的更新和保存

6、EUDAMED數(shù)據(jù)庫及UDI
1、歐盟EUDAMED數(shù)據(jù)庫
實現(xiàn)MDR目標的一個關鍵點是建立歐洲醫(yī)療器械數(shù)據(jù)庫 (EUDAMED),該數(shù)據(jù)庫希望整合不同的電子系統(tǒng)來集中整理和處理相關的信息。
這些信息包括: 市場上的器械和相關的經(jīng)營相關方(授權代表、進口商、經(jīng)銷商),符合性評估,公告機構,證書,臨床試驗,警戒和市場監(jiān)督。
EUDAMED數(shù)據(jù)庫的目的:
a) 公眾和衛(wèi)生保健專業(yè)人員更好地獲取信息, 提高總體透明度;
b) 避免多個上報的要求;
c) 加強成員國之間的協(xié)調(diào);
d) 簡化和促進經(jīng)營相關方, 公告機構或贊助者和成員國, 成員國之間以及歐盟委員會之間的信息流動。
EUDAMED數(shù)據(jù)庫由六大模塊組成,包括經(jīng)濟運營商注冊,唯一器械識別(UDI)和設備注冊,公告機構及證書,臨床調(diào)查和性能研究,警戒系統(tǒng)及上市后監(jiān)督,和市場監(jiān)督。目前EUDAMED 數(shù)據(jù)庫已開始使用三個模塊:參與者注冊、唯一器械識別(UDI)和設備注冊、公告機構和證書,在此提醒各位制造商,產(chǎn)品在銷往歐盟地區(qū)之前,應完成在EUDAMED中的產(chǎn)品注冊工作。其它模塊如臨床調(diào)查和性能研究、警戒系統(tǒng)和上市后監(jiān)督、市場監(jiān)督還沒有開始實施。
5、該區(qū)域有關UDI的要求
◆ Basic UDI-DI
Basic UDI-DI是歐盟MDR的特殊要求,當醫(yī)療器械產(chǎn)品同時具有以下相同特性時,則可以采用相同的Basic UDI-DI:
a) 預期目的
b) 風險等級
c) 基本設計和生產(chǎn)特性
企業(yè)根據(jù)產(chǎn)品的特性分配相應系列或家族產(chǎn)品的Basic UDI-DI。
Basic UDI-DI 不需要體現(xiàn)在產(chǎn)品標簽、包裝上,只需要體現(xiàn)在技術文檔、符合性聲明,并且在最終獲得的MDR EU證書上體現(xiàn)Basic UDI-DI。
◆ UDI
UDI 系統(tǒng)是醫(yī)療器械數(shù)據(jù)庫的一部分,目的是對醫(yī)療器械的識別提供一個全球協(xié)調(diào)化的框架。
UDI 是基于國際認可的器械標識和編碼標準創(chuàng)建的一系列數(shù)字或字母數(shù)字, 以便明確識別市場上的特定器械。
UDI 由 UDI-DI 和 UDI-PI 組成,必須標記在產(chǎn)品標簽、包裝或產(chǎn)品上。UDI-DI 必須登記在醫(yī)療器械數(shù)據(jù)庫中。

7、現(xiàn)場審核要求及注意事項
依據(jù)ISO 17021,MDR的現(xiàn)場體系審核分為兩個階段,stage 1 (階段1)及stage 2 (階段2)。而MDR (Regulation (EU) 2017/745)質(zhì)量體系要求是在EN ISO 13485:2016+A11:2021的基礎增加了法規(guī)的相關要求如警戒系統(tǒng),歐盟上市后監(jiān)督、上市后臨床跟蹤等。
8、臨床評價
1、為什么要進行臨床評價?
依據(jù)MDR章節(jié)VI第61條的要求,器械在正常條件下按照預期用途使用時,需要確認其符合MDR附錄I中規(guī)定的相關“基本安全和性能要求”(GSPRs),同時需評估MDR附錄I的第1節(jié)和第8節(jié)中提到的不良副作用和收益-風險比的可接受性,以上應基于提供充分臨床證據(jù)的臨床數(shù)據(jù),包括附錄III中提到的適用的相關數(shù)據(jù)。
制造商應詳細說明并證明必要的臨床證據(jù)的水平以證明符合相關的“基本安全和性能要求”(GSPRs)。臨床證據(jù)水平的選擇應與器械的特點及其預期用途相對應。
同時,制造商應按照MDR章節(jié)VI第61條和附錄XIV的a部分計劃、實施和記錄臨床評估。
2、基于“基本安全和性能要求”(GSPRs),臨床評價需收集充分的臨床證據(jù)用于:
a) 確認器械的預期用途,臨床受益,使用條件及禁忌癥。
b) 支持所有宣稱的器械預期的適應癥,預期的患者(特別是高風險人群),型號,附件,風險管理輸出的臨床風險點,殘余風險的可接受性。
c) 基于預期適用的患者和適應癥,確定當前技術水平下管理(包含治療或緩解或彌補等)該適應癥的臨床性能及臨床安全的最低可接受程度,該可接受程度范圍來源于當前技術水平下,其他技術管理(包含治療或緩解或彌補等)同類型患者和適應癥的程度的匯編。
基于行內(nèi)金標產(chǎn)品(Bench Mark),確定器械的基準范圍。
論證器械的臨床性能和安全,確認其受益-風險比在當前技術水平下,是可接受的。
◆ 臨床證據(jù)的定義
指與器械相關的臨床數(shù)據(jù)和臨床評價結(jié)果,其數(shù)量和質(zhì)量足以對器械是否安全以及在按制造商預期使用時是否達到預期臨床益處進行合格評估;
◆ 臨床數(shù)據(jù)
“臨床數(shù)據(jù)”是指器械使用過程中產(chǎn)生的有關安全性或性能的信息,來源如下:
- 器械自身的臨床試驗
- 等同器械的臨床試驗或在科學文獻中報告的其他研究
- 在同行評審的科學文獻中發(fā)表的關于器械或等同器械其他臨床經(jīng)驗的學術報告
- 來自上市后監(jiān)測的臨床相關信息,特別是上市后臨床隨訪
◆ 實質(zhì)性等同的定義
在進行臨床評估時,二類及以下非植入器械最常采用來源于等同器械的臨床數(shù)據(jù),需依據(jù)MDR附錄XIV的a部分第3條的要求從技術,生物學,臨床等三個方面進行實質(zhì)等同性論證。
有關以上實質(zhì)性等同三個方面具體內(nèi)容如下:
- 技術:器械的設計相似;在相似的使用條件下使用;具有相似的規(guī)格和性能,包括物理化學性能,如能量強度、抗拉強度、粘度、表面特性、波長和軟件算法;在相關的地方使用類似的部署方法;具有相似的操作原理和關鍵性能要求;
- 生物學:器械使用與相同人體組織或體液接觸的相同材料或物質(zhì),接觸的種類和持續(xù)時間相似,物質(zhì)的釋放特性相似,包括降解產(chǎn)物和可浸出物;
- 臨床:該裝置用于相同的臨床狀況或目的,包括相似的嚴重程度和疾病階段,在身體的同一部位,在相似的人群中,包括年齡、解剖學和生理學;具有相同類型的用戶;鑒于特定預期目的的預期臨床效果,具有類似的相關關鍵性能。
9、其他注意事項或特別提醒
1、產(chǎn)品帶CE標識銷售時,請使用最終通過審核的終版說明書和標簽。
2、關于抽樣的計劃:如果企業(yè)首次申請多個產(chǎn)品(限于IIa類、非植入或植入WET器械IIb類),公告機構一般為抽樣審核,但需要在一個證書周期內(nèi),審核完證書覆蓋的所有產(chǎn)品 。
MDR 法規(guī)提出了對患者及操作者更高的保護。它通過強調(diào)受益與危害的分析,明確可用性要求,強調(diào)充分的臨床證據(jù),明確要求制造商建立、完善并實施上市后監(jiān)督系統(tǒng)(尤其是主動式監(jiān)督)等方面來實現(xiàn)。

來源:廣東省醫(yī)療器械管理學會


