您當(dāng)前的位置:檢測資訊 > 科研開發(fā)
嘉峪檢測網(wǎng) 2024-05-31 11:39
摘要:雜質(zhì)作為藥品研發(fā)的一項(xiàng)關(guān)鍵質(zhì)量屬性,對藥物純度有著重要的影響,制定質(zhì)量控制策略,建立合理的檢查方法是研發(fā)過程中研究的重要內(nèi)容。
新原料藥:先前尚未在任何成員國或地區(qū)注冊的具有治療作用的活性成分(也稱為新分子或新化學(xué)實(shí)體)。它可以是某種已獲批準(zhǔn)的藥物的一種復(fù)合物、簡單的酯或鹽。
雜質(zhì)譜:對存在于某一新原料藥中的已鑒定或未鑒定雜質(zhì)的數(shù)量及含量。
原料藥的雜質(zhì)譜分析對應(yīng)CTD格式申報(bào)資料中的 M3.2.S.3.2雜質(zhì)(不考率安慰劑的情況下)。
本文主要結(jié)合作者幾年來的CTD資料撰寫的工作經(jīng)驗(yàn),依據(jù)ICH Q3A中對新原料藥的雜質(zhì)定義并結(jié)合案例對化學(xué)合成原料藥雜質(zhì)譜分析的一般原則和檢查方法的研究。
雜質(zhì)雜質(zhì)分類及來源:

根據(jù)ICH Q3C指導(dǎo)原則,對原料藥制備過程中引入的所有溶殘的測定方法進(jìn)行研究,并建立相應(yīng)的限度,各殘留溶劑的控制限度與ICH Q3C(R5)“雜質(zhì),殘留溶劑指導(dǎo)原則”推薦限度一致。
溶劑殘留的雜質(zhì)限度:
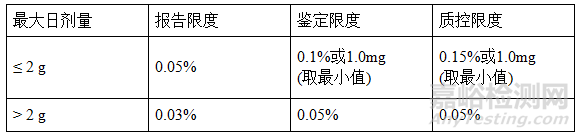
雜質(zhì)圖譜分析案例-經(jīng)驗(yàn)分享
使用1H-NMR 對內(nèi)標(biāo)進(jìn)行測定
如下圖所示;

該項(xiàng)目中選擇1,3,5-三甲氧基苯作為內(nèi)標(biāo),因?yàn)榭紤]到它的峰不會對雜質(zhì)A樣品中存在的任何峰造成干擾,并在CDCl3中制備樣品,使兩種材料都以等摩爾量的形式存在。

5秒是所有峰返回正值所需的最短時(shí)間。根據(jù)計(jì)算,其中T大約等于tnull/ln(2),新原料中雜質(zhì)(假設(shè)該雜質(zhì)為A)分析的時(shí)間T將為14秒。松弛延遲應(yīng)≥ (7/3)*T,相當(dāng)于大約17秒。建議增加5-10秒作為安全余量。因此,該方法中將延遲設(shè)置為30秒。
樣品溶液在t=0 時(shí)提交,測定結(jié)果為99.8%。20小時(shí)后重新提交相同的樣品,檢測結(jié)果為100.8%。由于差異≤ 2.0%,因此認(rèn)為樣品溶液可穩(wěn)定長達(dá)20小時(shí)。
該方法的運(yùn)行結(jié)果證明,該方法適用于1H-NMR 測定雜質(zhì)A。
HPLC法:
1.首先確定色譜條件,包括色譜柱的選擇(在合成工藝中,分析方法純化使用C18的硅膠柱比較多,僅針對于此雜質(zhì),色譜柱的選擇應(yīng)結(jié)合雜質(zhì)本身的理化特性,進(jìn)行篩選,對于相對穩(wěn)定的物質(zhì)多選用C18硅膠色譜柱),流動相的確定(根據(jù)分子本身的極性、溶解度等性質(zhì)進(jìn)行確定,實(shí)驗(yàn)室內(nèi)多用水和乙腈進(jìn)行分析純化),稀釋劑的選擇(稀釋劑盡可能的選擇與流動相溶劑相同,為了降低對設(shè)備的損耗或因溶劑的選擇而致使產(chǎn)品分析不純等情況)
2.溶液制備
3.圖譜分析
經(jīng)驗(yàn)分享:
以下通過三種分析方法對某一新原料藥中關(guān)于雜質(zhì)B純度進(jìn)行分析
方法一:等度洗脫

0.1%濃度水平色譜峰的信噪比為61。

樣品溶液在6.6分鐘處檢測到的峰可能是二氯-雜質(zhì)B,但需要通過保留標(biāo)記進(jìn)行確認(rèn)。在11.7分鐘處檢測到的峰是未知峰。

方法二:梯度

0.1%濃度水平色譜峰的信噪比為61。

樣品溶液的面積%數(shù)據(jù)與方法一一致。在11.4分鐘處檢測到的峰可能是二氯-雜質(zhì)B,但需要通過保留標(biāo)記進(jìn)行確認(rèn)。在13.8分鐘處檢測到的峰是未知峰。
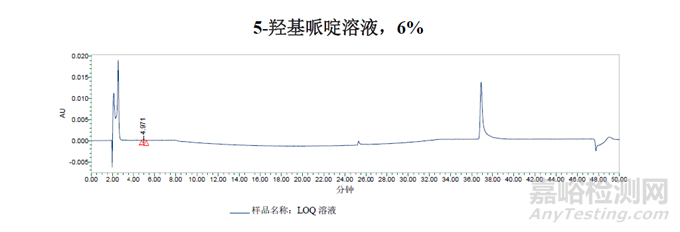
從5-羥基哌啶(6%)的稀溶液中獲得的峰似乎并未保留,而是隨進(jìn)樣峰被洗脫為切分峰。
通過對雜質(zhì)圖譜的分析得出的結(jié)論是:用于雜質(zhì)B純度測定的方法二并不適用于測定5-羥基哌啶。稀釋劑需要更高的含水量,以防止早期洗脫組分的峰切分。梯度也需要從含水量更高的條件開始,為了從進(jìn)樣峰中分離出5-羥基嘧啶峰。
方法三:自行進(jìn)行分析方法開發(fā)并經(jīng)過驗(yàn)證
對方法二進(jìn)行修改,通過更換流動相A,并同時(shí)改變稀釋劑的配比,增加稀釋劑的極性,從而防止峰切分。方法二中的其他條件保持不變。

使用含水量更高的條件,從進(jìn)樣峰中分離出5-羥基哌啶峰未觀察到切分峰。
結(jié)論:但是通過對方法三做出的修改似乎足以確定雜質(zhì)B的純度。
pH值測定法:
樣品將以溶劑(溶劑的選取應(yīng)是可以與水進(jìn)行互溶溶劑)和水層的雙相混合物形式提供。將樣品轉(zhuǎn)移至分液漏斗中,使各相分離。
將底部(水層)收集到適當(dāng)大小的燒杯中,并測量pH值和溫度。讀數(shù)應(yīng)記錄在20-25℃之間。
KF庫倫滴定法:
首先應(yīng)進(jìn)行溶液的配制,其次將配制好的樣品放置設(shè)備中進(jìn)行分析(應(yīng)多次進(jìn)樣進(jìn)行水分測定),根據(jù)公式計(jì)算水的含量,并確保重復(fù)檢測結(jié)果符合以下要求,< 0.10%不限,0.10-1.0%絕對誤差在0.2%以內(nèi),> 1.0%絕對誤差在0.5%以內(nèi)。
根據(jù)下列公式用儀器計(jì)算水含量:
含水量 = R-(B+(D ⅹ T))
式中:
水(R) = 消耗量(mC)/10.72(其中10.72 = mC,相當(dāng)于1μg水)
D = 偏移(μg/min)
T = 滴定時(shí)間(分鐘)
注冊申請中應(yīng)提供書面文件,證明分析方法是經(jīng)過驗(yàn)證并適用于雜質(zhì)的檢測和定量(可以參見ICH Q2A及Q2B分析方法論證指導(dǎo)原則項(xiàng)下)。技術(shù)因素(如生產(chǎn)能力與質(zhì)控方法)可作為部分依據(jù)來論證或選擇其他的雜質(zhì)限度。如果研發(fā)中所采用的分析方法和準(zhǔn)備上市產(chǎn)品的分析方法不同,在申報(bào)資料中應(yīng)予以討論。

來源:藥研


