鎵、鍺、銦作為典型的稀散金屬,很少有獨立礦床存在。鎵主要伴生在鋁土礦中,少量存在于錫礦、鎢礦、鉛鋅礦中。鍺多伴生在含硫化物的鉛、鋅、銅等礦物中或以含鍺褐煤存在。銦主要富集于硫化礦中,特別是閃鋅礦中,還有一部分伴生在方鉛礦、氧化鉛礦等礦石中。隨著鎵、鍺、銦需求量的增加,其戰略地位越來越突出,國家“十四五”科技創新重點研發計劃已經將鎵、鍺、銦等稀散金屬的開發利用列為重點專項。在冶煉鋅精礦時,鎵、鍺、銦會隨著硫化鋅礦石一起被浮選進入鋅精礦中,因此建立靈敏度高、分析快速、結果可靠的測定稀散金屬元素鎵、鍺、銦含量的方法已成為鋅精礦分析的重點。
目前,鎵、鍺、銦的常見測定方法有分光光度法、原子熒光光譜法、原子吸收光譜法等。但是,鋅精礦中稀散金屬元素含量比較低,上述方法需要進行分離、萃取、富集等過程,操作繁瑣、工作效率低,導致檢測結果的不確定度增加。電感耦合等離子體質譜法(ICP-MS)具有快速、靈敏度高、準確度好、檢出限低、動態線性范圍寬、可多元素同時測定等特點,在地質樣品稀散金屬元素檢測中的應用日益增多,包括在精礦中的應用。在國家標準GB/T 8151系列分析方法中,鍺測定方法規定的檢測范圍(質量分數)為0.0005%~0.10%,銦測定方法規定的檢測范圍(質量分數)為0.0020%~0.10%,無法檢出鋅精礦中低于0.0005%的鍺和低于0.0020%的銦,且缺乏鎵的測定方法。一些鋅精礦中鎵質量分數最高可達0.03%,具有較高的回收價值。因此,建立鋅精礦中鎵、鍺、銦同時測定的方法,對指導鋅精礦冶煉生產和回收利用等工作具有重要意義。
本文根據鋅精礦特性,采用硝酸-氫氟酸-高氯酸溶樣,同時滴加2滴25%(體積分數,下同)硫酸溶液,避免鐵氧化物析出。在進行ICP-MS分析時,探討了共存元素、試劑的干擾,并選擇內標元素103Rh校正71Ga、74Ge,187Re校正115In,同時以在線/離線公式校正74Se對74Ge以及115Sn對115In的干擾;當實際樣品中銅含量過高時,通過進一步稀釋來降低內標校正偏差。方法簡便、快捷,測定結果準確。
1、試驗方法
將鋅精礦樣品過篩后置于烘箱中加熱,冷卻備用。分取0.10g置于燒杯中,用少量水潤濕,加入硝酸蓋上表面皿,置于電熱板上,以不超過260℃低溫加熱至溶液體積為3~5mL,取下稍冷,加入氫氟酸、高氯酸和25%硫酸溶液,繼續加熱至白煙冒盡。如果消解液仍呈黑色,說明樣品含碳量較高,需要再加入高氯酸除盡有機物。取下稍冷,用水吹洗表面皿及燒杯壁,加入4mL50%(體積分數)硝酸溶液,蓋上表面皿,以不超過260℃低溫加熱溶解鹽類。取下冷卻,將上述溶液移入容量瓶中,用水稀釋至刻度,搖勻后用濾紙進行干過濾,棄去初濾液,收集中段濾液,供ICP-MS分析。其中,對于待測元素質量分數為0.00005%~0.0100%且銅質量分數小于8%的樣品,直接按照上述步驟測定;對于待測元素質量分數為0.00005% ~0.0100%且銅質量分數不小于8%的樣品,需要將濾液用2%硝酸溶液稀釋1倍后進行測定;對于待測元素質量分數為0.0100%~0.0500%的樣品,需要用2%硝酸溶液稀釋10倍后進行測定。同步進行空白試驗。
測定115In時,應同時測定115Sn,用公式(1)(ρSn/ρIn≤100的樣品,在線理論值校正)或公式 (2)(ρSn/ρIn>100的樣品,離線質量濃度校正)校正錫對銦的干擾;測定74Ge時,應同時測定77Se,用公式(3)校正硒對鍺的干擾。
結合各待測元素的標準曲線和上述公式對檢測結果進行校正和計算,所得結果減去空白值即為實際樣品溶液中待測元素的質量濃度。
2、結果與討論
2.1 稱樣量的選擇
樣品溶液中含有一定量的鹽,這可能引起基體效應,導致檢測結果出現偏差。雖然采用內標法可以校正鹽引起的漂移,但是如果鹽含量過大,內標校正的不確定度也同步增大。在滿足分析靈敏度的前提下,可以通過減少稱樣量的方法降低樣品溶液中鹽含量,但是稱樣量也不能過少,否則檢測結果易受儀器波動、試劑純度、器皿潔凈度等因素的影響。因此,試驗比較了稱樣量分別為0.05,0.10,0.15g時,20次連續測定所得待測元素測定值以及測定值的相對標準偏差(RSD)的變化,結果見表1。
表1 稱樣量的選擇(n=20)
由表1可知:稱樣量為0.05~0.15g時對待測元素測定值無顯著影響;稱樣量為0.10g時,測定值的RSD較低,同時各待測元素以及內標的信號強度降低率較低(<3%)。因此,試驗選擇的稱樣量為0.10g。
2.2 溶樣體系的選擇
在處理實際樣品時,既要保證樣品能完全分解,又要避免溶樣過程中待測元素揮發損失。溶樣時如果引入鹽酸,則會生成沸點分別為83,201℃的四氯化鍺和三氯化鎵,比較容易揮發,導致這兩種元素測定結果偏低。當樣品中這兩種待測元素含量較低時,上述影響可能不顯著。但是,一些鋅精礦中鍺質量分數高達0.05%,引入鹽酸可能造成較大的鍺損失,因此試驗不能采用鹽酸溶樣且需避免有氯環境。鎵、鍺、銦的硝酸鹽很穩定,不易揮發,因此試驗可選擇硝酸溶樣。考慮到鋅精礦中硫質量分數高達38%,用硝酸溶樣易形成硫球,導致待測元素被包裹,測定準確度變差,故采用加入高氯酸加熱至白煙冒盡的方法去除硫干擾。大部分鋅精礦中還含有硅化合物,不能被硝酸-高氯酸體系溶解,因此還需在溶樣體系中加入氫氟酸,以降低硅化合物夾雜的影響。約65%的鋅精礦中鐵質量分數大于8%,其中一些樣品中鐵的質量分數甚至高達20%,在溶樣結束后,鐵易形成不溶于硝酸的鐵氧化物,在樣品溶液中顯示為棕色渾濁物,影響測定結果和儀器壽命,試驗選擇加入2滴25%硫酸溶液來防止鐵氧化物的析出。為確定最優溶樣體系,以一個含二氧化硅、硫 、鐵質量分數分別為7.43%,33.5%,8.9%的樣品為待測對象,分別按如下5種體系溶樣,每種體系進行6次平行試驗,結果見表2。
表2 溶樣體系的選擇
由表2可知:以體系1#溶樣,鎵、鍺測定值較低,源于鹽酸引起的待測元素揮發;以體系2#溶樣,樣品溶液中有漂浮物,3種待測元素測定值略低,源于生成的包裹性硫球夾雜;以體系3#溶樣,樣品溶液底部有渣,3種待測元素測定值偏低,源于樣品中硅化合物夾雜;以體系4#溶樣,樣品溶液中產生了棕色渾濁物,源于生成的鐵氧化物;在體系4#中補加2滴25%硫酸溶液,即以體系5#溶樣時,樣品溶液變清亮,3種待測元素測定值較高。因此,試驗選擇采用體系5#溶樣。
2.3 待測元素同位素的選擇
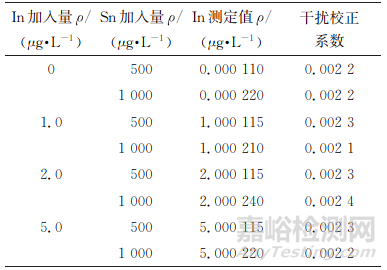
按照無干擾、豐度高的原則選擇各待測元素同位素。鎵可選的同位素有69Ga、71Ga,二者靈敏度接近,但考慮到138Ba2+豐度大(71.7%)且在鋅精礦中質量分數高達2.5%,對69Ga存在較大干擾,因此試驗選擇71Ga為待測元素鎵同位素。鍺可選的同位素有72Ge、73Ge、74Ge、76Ge,其中74Ge的靈敏度較高,但是74Ge會受同量異位素74Se的干擾,考慮到鋅精礦中硒質量分數普遍低于0.00005%,遠低于鍺,且74Se豐度低(0.9%),可通過校正公式(3)消除其干擾。銦可選的同位素有113In、115In,其中113In靈敏度低,受111Cd干擾大且不能通過公式校正消除;115In受同量異位素115Sn的干擾,但是鋅精礦樣品中錫質量分數通常低于 0.1%,且115Sn豐度小(0.36%),可按照公式(1)進行在線理論值校正得到準確結果。但是,針對個別錫質量分數較高(最高達0.3%)且銦含量較低(ρSn/ρIn>100)的樣品,在線理論值校正可能會導致銦測定出現偏差。分別取銦標準溶液(100μg·L-1)0,1.00,2.00,5.00mL各2份于一組100mL容量瓶中,加入不同量錫標準溶液,按照儀器工作條件測定,結果見表3。
表3 115Sn對115In的干擾校正系數
由表3可知,115Sn對115In的干擾校正系數為0.0021~0.0024,以其平均值(0.0022)建立公式(2)離線校正ρSn/ρIn>100的樣品中銦的測定值。
2.4 質譜干擾
樣品中的一些共存元素會形成多原子離子質譜干擾,因此試驗考察了鎳對74Ge的干擾。分別取1.00mL100μg·L-1鍺標準溶液、5.00mL1000μg·L-1鍺標準溶液各2份于一組100mL容量瓶中,加入不同量鎳標準溶液,按照儀器工作條件測定,結果見表4。
表4 58Ni16O對74Ge的干擾試驗
由表4可知:鎳的存在對鍺的準確測定干擾不大,可忽略。
一些質譜干擾為氧化物、多原子離子和同量異位素干擾等。對于鎵、鍺、銦,這些干擾主要來自氯、鈰、釹、鉬。氯會和等離子體中大量存在的氬、氧、氫結合,形成的71ArCl干擾71Ga,72Cl2干擾72Ge;鉬形成的氧化物98Mo17O干擾115In;142Ce2+ 、142Nd2+干擾71Ga。其中:本試驗不引入鹽酸,避免了氯干擾;鋅精礦中鈰、釹、鉬含量極微且干擾系數低,其干擾可以忽略不計。
2.5 非質譜干擾和內標的選擇
部分鋅精礦中氯質量分數達0.5%,為考察氯對鍺測定的影響,試驗取1.00,50.00mL100μg·L-1混合標準溶液B各2份于聚四氟乙烯燒杯中,分別加入不同量氯(滴加鹽酸),按照儀器工作條件測定,結果見表5。
表5 氯的干擾試驗
由表5可知,當氯加入量為0.5mg時,鍺回收率較理想,說明鋅精礦氯質量分數高達0.5%時也不會引起鍺測定偏差;當氯加入量為50mg (約2滴鹽酸)時,鍺測定結果明顯降低,可能源于鍺揮發損失,因此試驗應避開含氯環境,同時不應引入含氯試劑。
分析某鋅精礦企業近3年的檢測數據可知,鋅精礦中主量成分分布(質量分數中位值)為鋅62%、鉛10%、鐵20% (約65%的樣品中鐵質量分數大于8%)、硫38% 、二氧化硅13%、鈣10%、銅8% (約95%的樣品中銅低于質量分數 2%)、鎘1%、鋁5%、鎳 5%、鈷 5%、錳 5%、鋇 2.5%、硒<0.00005%,部分成分含量較高,可能影響待測元素的準確測定。試驗選擇采用內標法消除主量成分的干擾,內標元素同位素按照豐度高、無干擾,質量數與待測元素相近,樣品中不含內標元素的原則來選擇。鋅精礦普查數據(中位值)顯示,鋅精礦中鑭質量分數約0.0006%,釔質量分數約0.0005%,鈧、銠、銥、錸的質量分數均小于 0.000001%,試驗選擇以45Sc、103Rh、187Re作內標元素,并考察了主要共存干擾元素鉛、鋅、銅對待測元素和內標元素的影響。在1.00,50.00μg·L-1混合標準溶液(內標元素的質量濃度為25.00μg·L-1)200mL中分別加入62mgZn、10mgPb、2mgCu、4mgCu、8mgCu以及62mgZn+10mgPb+4mgCu,按照儀器工作條件測定,結果見表6。
表6 主量元素干擾試驗
由表6可知:當存在50mg·L-1Pb、40mg·L-1Cu時,鎵、鍺、銦測定結果較高,其中鎵測定結果偏高較多,其他共存元素干擾不大;鈧回收率稍高于待測元素的,銠的回收率和鎵、鍺的基本一致,錸的回收率和銦的基本一致,因此試驗選擇以103Rh校正71Ga、74Ge、187Re校正115In。但是,當銅質量濃度不小于40mg·L-1(銅質量分數不小于8%)時,內標法校正結果有一定偏差,應進一步稀釋樣品溶液,使銅質量濃度小于40mg·L-1后再進行上機測定。
2.6 試劑的干擾
試驗引入的試劑,如硝酸影響較小,氫氟酸和高氯酸在測定前已被加熱損失掉,少量硫酸可能影響待測元素的準確測定,因此試驗考察了硫酸的干擾。在1.00,50.00μg·L-1混合標準溶液100mL中分別加入1~3滴25%硫酸溶液,按照儀器工作條件測定。結果顯示,不加內標時3種待測元素的回收率為97.0%~106%,經內標校正后各待測元素的回收率接近100%,說明微量硫酸的存在不干擾待測元素的測定。
2.7 標準曲線和檢出限
按照儀器工作條件測定混合標準溶液系列,以各待測元素的質量濃度為橫坐標,對應的信號強度為縱坐標繪制標準曲線。結果顯示,各待測元素標準曲線的線性范圍均為0.20%~50.00μg·L-1,線性回歸方程和相關系數見表7。
按照試驗方法制備11份樣品空白溶液,在選定的儀器工作條件下測定各元素含量,計算各待測元素測定值的標準偏差(s),以3s作為各元素的檢出限,結果見表7。
表7 線性參數和檢出限
2.8 標準物質和樣品分析
按照試驗方法分析標準物質 GBW 07168和兩個鋅精礦樣品(編號分別為2#和3#),各平行測定11次,計算測定值的 RSD,并對這兩個樣品進行加標回收試驗,計算回收率;同時參考文獻分析樣品2#和3#。所得結果見表8。
表8 精密度和準確度試驗結果(n=11)
由表8可知:標準物質中3種待測元素的測定值和認定值基本一致,實際樣品中3種待測元素的測定值和文獻方法的測定值也基本一致,實際樣品中3種待測元素的回收率為95.0%~106%,說明方法的準確度較好;測定值的RSD均 不大于8.5%,說明方法的精密度較好,滿足鋅精礦中鎵、鍺、銦的測定要求。
3、試驗結論
本文采用硝酸-氫氟酸-高氯酸溶樣體系分解樣品,并在待測樣品溶液中加入2滴25%硫酸溶液避免鐵氧化物的析出;采用ICP-MS測定樣品溶液中鎵、鍺、銦的含量,同時探討了待測/內標元素同位素的選擇以及質譜、非質譜和試劑干擾及消除。該方法簡便、快捷,能用于鋅精礦中鎵、鍺、銦的測定。
作者:譚秀麗1,左鴻毅2*,王文杰1
單位:1.深圳市中金嶺南有色金屬股份有限公司韶關冶煉廠;
2.深圳市中金嶺南有色金屬股份有限公司科學技術開發院
來源:《理化檢驗-化學分冊》2024年第2期