前言
2023年7月3日,CDE官網(wǎng)發(fā)布了《化學(xué)原料藥受理審查指南(試行)》(2023年第38號)。這是首次發(fā)布專門針對原料藥的受理審查指南,不像之前都是“鑲嵌”在《藥品注冊受理審查指南》中,制劑和原料藥雜糅在一起,需要費(fèi)工夫精挑細(xì)選出原料藥適用的注冊要求。如今,響應(yīng)2020年《藥品注冊管理辦法》中關(guān)于化學(xué)原料藥單獨(dú)審評審批的程序,在2020-04-30和2022-02-09相繼發(fā)布了兩版征求意見稿之后,正式版的化學(xué)原料藥受理審查指南終于姍姍來遲。
這次的受理審查指南是“自發(fā)布之日起施行”,真是連一點(diǎn)點(diǎn)適應(yīng)和準(zhǔn)備的時間都不給我們。于是,離提交登記資料就差一步的RA們肯定想快速了解,相比于之前的受理要求,什么變了,什么沒有變,然后主攻“差異點(diǎn)”,盡快補(bǔ)足相關(guān)內(nèi)容,追趕deadline。那么,下面就從五個方面來梳理和匯總下最新指南相較于2020年7月發(fā)布《化學(xué)藥品注冊受理審查指南(試行)》中原料藥受理審查要求做出了哪些更新。
友情提示:如果沒時間仔細(xì)看中間的大段對比內(nèi)容,可直接跳到文后的8個要點(diǎn)總結(jié)。
一、 適用范圍
無實質(zhì)變化。對于原料藥,均適用于上市申請。
|
適用范圍 |
|
2020-07《化學(xué)藥品注冊受理審查指南(試行)》 |
2023-07《化學(xué)原料藥受理審查指南(試行)》 |
|
化學(xué)藥品(注冊分類1、2、3、4、5.1、5.2類)的藥物臨床試驗申請/藥品上市許可申請。 |
化學(xué)原料藥上市申請登記。 |
二、 受理部門
無變化。無論是境內(nèi)還是境外生產(chǎn)原料藥,受理部門仍為CDE。
|
受理部門 |
|
2020-07《化學(xué)藥品注冊受理審查指南(試行)》 |
2023-07《化學(xué)原料藥受理審查指南(試行)》 |
|
由國家藥品監(jiān)督管理局藥品審評中心受理。 |
國家藥品監(jiān)督管理局藥品審評中心。 |
三、 資料基本要求
總體要求中,在參考的法規(guī)指南中,新增了《國家藥監(jiān)局關(guān)于進(jìn)一步完善藥品關(guān)聯(lián)審評審批和監(jiān)管工作有關(guān)事宜的公告》,并強(qiáng)調(diào)“登記資料文件夾目錄按要求進(jìn)行編制”。
|
資料基本要求--總體要求 |
|
2020-07《化學(xué)藥品注冊受理審查指南(試行)》 |
2023-07《化學(xué)原料藥受理審查指南(試行)》 |
|
按照《藥品注冊管理辦法》及《化學(xué)藥品注冊分類及申報資料 要求》的規(guī)定,提供符合要求的申報資料。申報資料應(yīng)根據(jù)現(xiàn)行版《M4:人用藥物注冊申請通用技術(shù)文檔(CTD)》格式整理,目錄及項目編號不能改變,對應(yīng)項目無相關(guān)信息或研究資料,項目編號和名稱也應(yīng)保留,可在項下注明“不適用”并說明理由。 |
按照《藥品注冊管理辦法》、《國家藥監(jiān)局關(guān)于進(jìn)一步完善藥品關(guān)聯(lián)審評審批和監(jiān)管工作有關(guān)事宜的公告》、《化學(xué)藥品注冊分類及申報資料要求》的規(guī)定,提供符合要求的登記資料。登記資料應(yīng)根據(jù)現(xiàn)行版《M4:人用藥物注冊申請通用技術(shù)文檔(CTD)》格式整理,目錄及項目編號不能改變。登記資料文件夾目錄按要求進(jìn)行編制。 |
3.1 登記表
本次將《原料藥登記表》單獨(dú)列明,登記表填寫應(yīng)符合原料藥登記平臺上的填表說明,并確保遞交版登記表與在線提交的登記表的核對碼一致。特別強(qiáng)調(diào)這一點(diǎn),大概是因為每次修改登記表后,打印出來的登記表核對碼都會變化,核對碼相當(dāng)于登記表的身份證編號,具有唯一性,核對碼一致就等于內(nèi)容完全一致。
另外,無需提交《申報資料自查表》。
|
登記表 |
|
2020-07《化學(xué)藥品注冊受理審查指南(試行)》 |
2023-07《化學(xué)原料藥受理審查指南(試行)》 |
|
藥品注冊申請表、申報資料自查表、小型微型企業(yè)收費(fèi)優(yōu)惠申請表(如適用)與申報資料份數(shù)一致,其中至少一份為原件;填寫應(yīng)當(dāng)準(zhǔn)確、完整、規(guī)范,不得手寫或涂改,并應(yīng)符合填表說明的要求。 |
《原料藥登記表》,《小型微型企業(yè)收費(fèi)優(yōu)惠申請表》(如適用)填寫應(yīng)當(dāng)準(zhǔn)確、完整、規(guī)范,不得手寫或涂改,并應(yīng)符合填表說明的要求。登記表數(shù)據(jù)核對碼應(yīng)與在線提交的登記表核對碼一致。 |
|
各頁的數(shù)據(jù)核對碼必須一致,并與提交的電子申請表一致,申請表及自查表各頁邊緣應(yīng)加蓋申請人或注冊代理機(jī)構(gòu)騎縫章。 |
(作者注:《原料藥申報登記資料提交說明》要求登記表打印表格各頁邊緣應(yīng)當(dāng)騎縫加蓋負(fù)責(zé)辦理登記事宜機(jī)構(gòu)或者藥品注冊代理機(jī)構(gòu)的公章。) |
3.2 登記資料的整理
2020年版的要求適用于制劑資料的紙質(zhì)遞交,在鼓勵全面電子遞交的當(dāng)下早已過時。《國家藥監(jiān)局關(guān)于實施藥品注冊申請電子申報的公告》(2022年第110號)明確要求:“自2023年1月1日起,申請人提交的國家藥監(jiān)局審評審批藥品注冊申請以及審評過程中補(bǔ)充資料等,調(diào)整為以電子形式提交申報資料,申請人無需提交紙質(zhì)申報資料”。《關(guān)于藥品注冊申請電子申報有關(guān)要求的通知》規(guī)定“原料藥登記資料應(yīng)采用PDF格式文件進(jìn)行整理,電子簽章等相關(guān)要求應(yīng)符合申報資料電子光盤技術(shù)要求”,并提供了3個附件:申報資料電子光盤技術(shù)要求、藥品注冊申請電子文檔結(jié)構(gòu)、承諾書模板。
|
登記資料的整理 |
|
2020-07《化學(xué)藥品注冊受理審查指南(試行)》 |
2023-07《化學(xué)原料藥受理審查指南(試行)》 |
|
2套完整申請資料(至少1套為原件)+1套綜述資料(應(yīng)包含模塊一、模塊二),每套裝入相應(yīng)的申請表及目錄。 |
登記人應(yīng)按照《國家藥監(jiān)局關(guān)于實施藥品注冊申請電子申報的公告》(2022年第110號)、《關(guān)于藥品注冊申請電子申報有關(guān)要求的通知》的要求準(zhǔn)備電子申報資料,登記人需對申報資料中的所有PDF文件使用登記人或注冊代理機(jī)構(gòu)的電子簽章。 |
|
除《藥品注冊申請表》及檢驗機(jī)構(gòu)出具的檢驗報告外,申報資料(含圖譜)應(yīng)逐個封面加蓋申請人或注冊代理機(jī)構(gòu)公章,封面公 章應(yīng)加蓋在文字處,整理規(guī)范詳見《藥品注冊申報資料格式體例與 整理規(guī)范》。 |
四、 形式審查要點(diǎn)
4.1 光盤及文件格式審查要點(diǎn)
其實原料藥通過光盤提交電子資料已經(jīng)有好多年了。《關(guān)于藥品注冊申請電子申報有關(guān)要求的通知》要求“申請人需按本通知要求提交1套完整的電子申報資料光盤供審評使用”“實施電子申報后受理的藥品注冊申請,審評過程中補(bǔ)充資料等采用電子申報資料形式進(jìn)行遞交”。
|
光盤及文件格式 |
|
2020-07《化學(xué)藥品注冊受理審查指南(試行)》 |
2023-07《化學(xué)原料藥受理審查指南(試行)》 |
|
NA |
光盤外觀清潔無劃痕并保持完整,光盤表面不應(yīng)粘貼標(biāo)簽。光盤內(nèi)容可順利讀取。光盤硬盒及檔案袋均按要求加貼封面。光盤中登記資料格式,文件夾及文件名符合《關(guān)于藥品注冊申請電子申報有關(guān)要求的通知》的要求。 |
4.2 登記事項審查要點(diǎn)
本次的登記事項審查要點(diǎn)十分清晰地解答了原料藥登記中的幾大經(jīng)典疑問:
(1)制劑中間體能不能登記?答曰:不能。
(2)不同生產(chǎn)工藝能否登記在同一個號下?不同質(zhì)量標(biāo)準(zhǔn)能否登記在同一個號下?不同生產(chǎn)場地能否登記在同一個號下?答曰:都不可以。
(3)那同一企業(yè)生產(chǎn)的同一名稱原料藥可否同時登記多個號?答曰:可以試試,但需要提供充分理由(比如生產(chǎn)線、生產(chǎn)工藝等有所不同)。
(4)新登記的原料藥技術(shù)審評期間發(fā)生變更怎么辦,可以在審評期間遞交變更嗎?答曰:重大變更的、涉及技術(shù)審評內(nèi)容的都不可以,只能撤回等變更完再重新登記;其他的比如變更公司名稱、注冊地址的需要及時告知CDE并提交相關(guān)證明性資料。
|
登記事項審查要點(diǎn) |
|
2020-07《化學(xué)藥品注冊受理審查指南(試行)》 |
2023-07《化學(xué)原料藥受理審查指南(試行)》 |
|
NA |
1.登記人應(yīng)當(dāng)按照關(guān)聯(lián)審評審批制度要求,在化學(xué)原料藥、輔料及直接接觸藥品的包裝材料和容器登記平臺登記化學(xué)原料藥(不含制劑中間體)。 |
|
2.同一企業(yè)在同一生產(chǎn)場地生產(chǎn)的同一化學(xué)原料藥,生產(chǎn)工藝和質(zhì)量標(biāo)準(zhǔn)相同的,應(yīng)按照同一登記號登記。對同一原料藥存在不同生產(chǎn)工藝的,原則上,應(yīng)按照不同登記號進(jìn)行登記,并提交相應(yīng)登記資料。同一企業(yè)以不同登記號登記相同名稱化學(xué)原料藥的應(yīng)在登記表特別聲明事項中說明理由及原登記號的情況。 |
|
3.化學(xué)原料藥審評期間,發(fā)生可能影響藥品安全性、有效性和質(zhì)量可控性的重大變更的,登記人應(yīng)當(dāng)撤回原登記,補(bǔ)充研究后重新申報。登記人名稱變更、注冊地址名稱變更等不涉及技術(shù)審評內(nèi)容的,應(yīng)當(dāng)及時書面告知藥品審評中心并提交相關(guān)證明性資料。 |
|
4.境外生產(chǎn)化學(xué)原料藥登記人應(yīng)委托中國境內(nèi)的企業(yè)法人進(jìn)行登記,境內(nèi)生產(chǎn)化學(xué)原料藥的登記人應(yīng)為原料藥生產(chǎn)企業(yè)。 |
4.3 登記表審查要點(diǎn)
目前原料藥登記平臺已經(jīng)趨近成熟,從實操層面來看,登記表的填寫幾乎沒什么變化。在填寫登記表時,需要選擇關(guān)聯(lián)審評或單獨(dú)審評程序,單獨(dú)審評仍只適用于仿制境內(nèi)已上市藥品所用的化學(xué)原料藥。另外需要注意的一點(diǎn)是,未列入國家藥品標(biāo)準(zhǔn)或者藥品注冊標(biāo)準(zhǔn)的,需要提交通用名稱證明文件或同時提出通用名稱核準(zhǔn)申請(需單獨(dú)準(zhǔn)備光盤)。
|
2020-07《化學(xué)藥品注冊受理審查指南(試行)》 |
2023-07《化學(xué)原料藥受理審查指南(試行)》 |
|
1.藥品注冊申請表(注:該申請表只適用于制劑申報,此處略)2.小型微型企業(yè)收費(fèi)優(yōu)惠申請表如符合小微企業(yè)行政事業(yè)性收費(fèi)優(yōu)惠政策,可提交小型微型企業(yè)收費(fèi)優(yōu)惠申請表并提供如下信息:2.1 基本信息:如企業(yè)名稱、聯(lián)系人、聯(lián)系電話等,應(yīng)與藥品注冊申請表有關(guān)信息一致。2.2 從業(yè)人員、上一納稅年度營業(yè)收入、企業(yè)資產(chǎn)總值等:申請人依實際情況填寫,2.3 應(yīng)由其法定代表人或接受其授權(quán)者(另需提供簽字授權(quán)書原件)在此簽名、加蓋機(jī)構(gòu)公章(須與其機(jī)構(gòu)名稱完全一致)。 |
按照《原料藥登記表填表說明》的要求規(guī)范填寫登記表,填報信息應(yīng)與登記資料中相應(yīng)內(nèi)容保持一致。1.其他特別申明事項:如已向中檢院或省、自治區(qū)、直轄市藥品監(jiān)督管理部門申請前置注冊檢驗的,應(yīng)予以說明并提交送檢憑證。如符合小微企業(yè)免收注冊費(fèi)用的,登記人應(yīng)予以說明,并提交《小型微型企業(yè)收費(fèi)優(yōu)惠申請表》等電子資料。如屬于關(guān)聯(lián)審評審批情形的,登記人應(yīng)填寫被關(guān)聯(lián)制劑品種名稱及其申請人信息。2.上市申請登記時如屬于仿制境內(nèi)已上市藥品所用的化學(xué)原料藥可選擇單獨(dú)審評程序,并同時填寫境內(nèi)已上市制劑信息(至少填寫一個制劑)。3.產(chǎn)品中文名稱:仿制化學(xué)原料藥應(yīng)當(dāng)使用國家藥品標(biāo)準(zhǔn)或者藥品注冊標(biāo)準(zhǔn)收載的藥品通用名稱。未列入國家藥品標(biāo)準(zhǔn)或者藥品注冊標(biāo)準(zhǔn)的,登記人在提出上市申請登記時,應(yīng)當(dāng)提交通用名稱證明文件,或同時提出通用名稱核準(zhǔn)申請(需單獨(dú)準(zhǔn)備光盤)。4.包裝:如有多個包裝材質(zhì)要分別填寫,中間用句號分開。包裝規(guī)格:原則上應(yīng)使用具體明確的數(shù)字單位進(jìn)行表示,每一份登記表可填寫多個包裝規(guī)格。5.擬用制劑給藥途徑應(yīng)按照藥品實際情況準(zhǔn)確填寫,可多選。6.有效期:以月為單位填寫。如有多個包裝材質(zhì),有效期如有不同則要分別對應(yīng)填寫。7.是否涉及特殊管理藥品或成分:屬于麻醉藥品、精神藥品、醫(yī)療用毒性藥品、放射性藥品的,應(yīng)填寫。8.包材來源應(yīng)填寫包材名稱、登記號(如有)和生產(chǎn)商信息。9.化學(xué)原料藥登記人、生產(chǎn)企業(yè)、注冊代理機(jī)構(gòu)企業(yè)名稱、地址等信息應(yīng)與證明文件中相應(yīng)內(nèi)容保持一致,并指定其中一個申請機(jī)構(gòu)負(fù)責(zé)繳納注冊費(fèi)用。已經(jīng)填入的各機(jī)構(gòu)均應(yīng)當(dāng)由其法定代表人或接受其授權(quán)者(另需提供簽字授權(quán)書原件,授權(quán)書應(yīng)加蓋公章(如有))在此簽名、加蓋機(jī)構(gòu)公章(須與其機(jī)構(gòu)名稱完全一致)。 |
4.4 登記資料審查要點(diǎn)
從證明性文件的類型看,沒有什么變化,依然是老幾樣:包材來源和授權(quán)書、專利聲明、上市證明、GMP符合性證明、委托文書、營業(yè)執(zhí)照等。境外的官方出具證書需要公證認(rèn)證,境外登記人的委托文書需要公證。境外上市證明依然是CPP、FSC、CEP或DMF號(注:使用DMF號時的組合比較復(fù)雜)等。
需要特別注意的一點(diǎn)是,首次登記需要提交“工藝驗證方案和報告”。這在之前的80號文甚至M4中都未明確要求過(只對無菌原料藥有特殊要求),也算是新的中國特色了。對于境外生產(chǎn)的原料藥登記來說,翻譯量又加大了。
另外,仿制化學(xué)原料藥至少需要包括三個注冊批樣品6個月穩(wěn)定性試驗數(shù)據(jù)。
|
登記資料審查要點(diǎn) |
|
2020-07《化學(xué)藥品注冊受理審查指南(試行)》 |
2023-07《化學(xué)原料藥受理審查指南(試行)》 |
|
1.產(chǎn)品相關(guān)證明性文件 |
1.證明性文件 |
|
1.1 原料藥、藥用輔料及藥包材證明文件1.1.1 原料藥、藥用輔料及藥包材合法來源證明文件,包括供貨協(xié)議、發(fā)票等(適用于制劑未選用已登記原輔包情形)。1.1.2 原料藥、藥用輔料及藥包材授權(quán)使用書復(fù)印件(適用于制劑選用已登記原輔包情形)。如為供應(yīng)商出具,需有原料藥、藥用輔料和藥包材企業(yè)授權(quán),并附授權(quán)信復(fù)印件。1.2 專利信息及證明文件申請的藥物或者使用的處方、工藝、用途等專利情況及其權(quán)屬狀態(tài)說明,以及對他人的專利不構(gòu)成侵權(quán)的聲明。應(yīng)由申請人出具,并承諾對可能的侵權(quán)后果承擔(dān)全部責(zé)任。1.3 麻醉藥品、精神藥品研制立項批復(fù)文件復(fù)印件(如適用)1.4 對照藥來源證明文件(如適用)1.9 允許藥品上市銷售證明文件(適用于境外已上市的藥品)1.9.1 境外藥品管理機(jī)構(gòu)出具的允許該藥品上市銷售的證明文件、公證認(rèn)證文書及中文譯文.19.2 對于生產(chǎn)國家或地區(qū)按食品或按醫(yī)療器械管理的制劑,應(yīng)提供該國家或者地區(qū)有關(guān)管理機(jī)構(gòu)允許該品種上市銷售的證明文件。1.10 境外藥品管理機(jī)構(gòu)出具的允許藥品變更的證明文件、公證認(rèn)證文書及中文譯文(如適用)。2.申請人/生產(chǎn)企業(yè)證明性文件2.1 申請人資質(zhì)證明文件2.1.1 境內(nèi)申請人機(jī)構(gòu)合法登記證明文件(營業(yè)執(zhí)照等)、相應(yīng)的藥品生產(chǎn)許可證及其變更記錄頁(適用于上市許可申請)。2.1.2 境外申請人指定中國境內(nèi)的企業(yè)法人辦理相關(guān)藥品注冊事項的,應(yīng)當(dāng)提供委托文書、公證文書及中文譯文,以及注冊代理機(jī)構(gòu)的營業(yè)執(zhí)照復(fù)印件。上市許可申請時,如變更注冊代理機(jī)構(gòu),還應(yīng)提交境外申請人解除原委托代理注冊關(guān)系的文書、公證文書及中文譯文。2.2 生產(chǎn)企業(yè)資質(zhì)證明文件2.2.1 境內(nèi)生產(chǎn)企業(yè)機(jī)構(gòu)合法登記證明文件(營業(yè)執(zhí)照等)、相應(yīng)的藥品生產(chǎn)許可證及變更記錄頁(適用于上市許可申請)。2.2.2 境外藥品管理機(jī)構(gòu)出具的該藥品生產(chǎn)廠和包裝廠符合藥品生產(chǎn)質(zhì)量管理規(guī)范的證明文件、公證認(rèn)證文書及中文譯文(適用于境外生產(chǎn)藥品)。對于生產(chǎn)國家或地區(qū)按食品管理的制劑,應(yīng)提供該國家或地區(qū)藥品管理機(jī)構(gòu)出具的該生產(chǎn)企業(yè)符合藥品生產(chǎn)質(zhì)量管理規(guī)范的證明文件,或有關(guān)機(jī)構(gòu)出具的該生產(chǎn)企業(yè)符合ISO9000質(zhì)量管理體系的證明文件。對于生產(chǎn)國家或地區(qū)按醫(yī)療器械管理的制劑,應(yīng)提供企業(yè)資格證明文件。2.3 小微企業(yè)申報資料(如適用):企業(yè)的工商營業(yè)執(zhí)照副本復(fù)印件;上一年度企業(yè)所得稅納稅申報表(須經(jīng)稅務(wù)部門蓋章確認(rèn))或上一年度有效統(tǒng)計表(統(tǒng)計部門出具)原件。 |
1.1 藥包材證明文件1.1.1 藥包材合法來源證明文件,包括供貨協(xié)議、發(fā)票等(適用于原料藥未選用已登記包材情形)。1.1.2 藥包材授權(quán)使用書(適用于原料藥選用已登記包材情形)。如為供應(yīng)商出具,需有藥包材企業(yè)授權(quán),并附授權(quán)信。1.2 專利信息及證明文件登記原料藥的化合物、工藝、用途等專利情況及其權(quán)屬狀態(tài)說明,以及對他人的專利不構(gòu)成侵權(quán)的聲明。應(yīng)由登記人出具,并承諾對可能的侵權(quán)后果承擔(dān)全部責(zé)任。1.3 特殊藥品研制立項批準(zhǔn)文件(如適用)麻醉藥品和精神藥品需提供研制立項批復(fù)文件。1.4 境內(nèi)生產(chǎn)化學(xué)原料藥證明文件生產(chǎn)企業(yè)的營業(yè)執(zhí)照和已取得相應(yīng)范圍的《藥品生產(chǎn)許可證》及變更記錄頁。1.5 境外生產(chǎn)化學(xué)原料藥證明文件1.5.1 境外藥品管理機(jī)構(gòu)出具的允許該化學(xué)原料藥上市銷售證明文件、公證認(rèn)證文書及中文譯文,化學(xué)原料藥生產(chǎn)企業(yè)符合藥品生產(chǎn)質(zhì)量管理規(guī)范的證明文件、公證認(rèn)證文書及中文譯文。也可提供歐洲藥典適用性證明文件(CEP)與附件;或者該化學(xué)原料藥主控系統(tǒng)文件(DMF)的文件號以及采用該化學(xué)原料藥的制劑已在國外獲準(zhǔn)上市的證明文件及該原料藥生產(chǎn)企業(yè)符合藥品生產(chǎn)質(zhì)量管理規(guī)范的證明文件(若因該原料藥僅供企業(yè)集團(tuán)內(nèi)部使用等原因,而無法提供DMF號,可由境外登記人出具相應(yīng)情況說明)。對于生產(chǎn)國家或地區(qū)按食品管理的化學(xué)原料藥,應(yīng)提供該國家或地區(qū)藥品管理機(jī)構(gòu)出具的該生產(chǎn)企業(yè)符合藥品生產(chǎn)質(zhì)量管理規(guī)范的證明文件,或有關(guān)機(jī)構(gòu)出具的該生產(chǎn)企業(yè)符合ISO9000質(zhì)量管理體系的證明文件,和該國家或者地區(qū)有關(guān)管理機(jī)構(gòu)允許該品種上市銷售的證明文件。境外生產(chǎn)的1類、2.1類化學(xué)藥品所用原料藥參照相應(yīng)制劑要求提交相關(guān)證明文件。登記人應(yīng)承諾:審評審批期間,相關(guān)證明性文件載明的境外監(jiān)管狀態(tài)信息,如生產(chǎn)上市情況、GMP合規(guī)情況等內(nèi)容發(fā)生變化的,及時如實告知監(jiān)管部門。1.5.2 境外登記人指定中國境內(nèi)的企業(yè)法人辦理相關(guān)化學(xué)原料藥登記事項的,應(yīng)當(dāng)提供委托文書、公證文書及其中文譯文,以及注冊代理機(jī)構(gòu)的營業(yè)執(zhí)照。1.6 小微企業(yè)證明文件(如適用)企業(yè)的工商營業(yè)執(zhí)照副本復(fù)印件;上一年度企業(yè)所得稅納稅申報表(須經(jīng)稅務(wù)部門蓋章確認(rèn))或上一年度有效統(tǒng)計表(統(tǒng)計部門出具)原件。2.其他登記資料上市申請登記時應(yīng)按要求提交工藝驗證方案和報告。仿制化學(xué)原料藥至少需要包括三個注冊批樣品6個月穩(wěn)定性試驗數(shù)據(jù)。 |
4.5 其他提示
相同點(diǎn):符合WHO推薦格式的證明性文件,無需公證認(rèn)證;補(bǔ)正限期三十日(工作日)。
不同點(diǎn):“不適用的文檔清單及說明”需要在模塊一說明函中列明;審評審批過程中保持證明性文件始終在有效期內(nèi),過期需更新;光盤中需附《承諾書》;正當(dāng)理由逾期不予補(bǔ)正的,原資料由CDE直接銷毀,不再退回。
|
其他提示 |
|
2020-07《化學(xué)藥品注冊受理審查指南(試行)》 |
2023-07《化學(xué)原料藥受理審查指南(試行)》 |
|
1.麻醉藥品、精神藥品、醫(yī)療用毒性藥品、藥品類易制毒化學(xué)品不得委托生產(chǎn);但是,國務(wù)院藥品監(jiān)督管理部門另有規(guī)定的除外。 |
1.模塊一說明函內(nèi)容應(yīng)同時提交不適用的文檔清單及說明(如適用)。 |
|
2,化學(xué)原料藥、藥用輔料及藥包材與藥品制劑關(guān)聯(lián)審評審批要求,參照《藥品注冊管理辦法》及相關(guān)規(guī)定辦理。在藥品制劑注冊申請時,由藥品制劑注冊申請人一并提供原料藥研究資料的,應(yīng)嚴(yán)格按照要求提交完整的原料藥申報資料。3.境外生產(chǎn)的藥品所提交的境外藥品管理機(jī)構(gòu)出具的證明文件(包括允許藥品上市銷售證明文件、符合藥品生產(chǎn)質(zhì)量管理規(guī)范證明文件以及允許藥品變更證明文件等),為符合世界衛(wèi)生組織推薦的統(tǒng)一格式原件的,可不經(jīng)所在國公證機(jī)構(gòu)公證及駐所在國中國使領(lǐng)館認(rèn)證。5.申請人應(yīng)當(dāng)在三十日內(nèi)完成補(bǔ)正資料,申請人無正當(dāng)理由逾期不予補(bǔ)正的,視為放棄申請,并將申報資料退回給申請人。 |
2.境外生產(chǎn)的化學(xué)原料藥所提交的境外藥品管理機(jī)構(gòu)出具的證明文件(包括允許化學(xué)原料藥上市銷售證明文件、符合藥品生產(chǎn)質(zhì)量管理規(guī)范證明文件),為符合世界衛(wèi)生組織推薦的統(tǒng)一格式的,可不經(jīng)所在國公證機(jī)構(gòu)公證及駐所在國中國使領(lǐng)館認(rèn)證。3.審評審批過程中,如相關(guān)證明性文件超過有效期,申請人應(yīng)當(dāng)及時通過公文向藥審中心遞交更新的證明性文件。4.光盤資料中應(yīng)按照《關(guān)于藥品注冊申請電子申報有關(guān)要求的通知》的要求提供承諾書。5.登記人應(yīng)當(dāng)在三十日內(nèi)完成補(bǔ)正資料,登記人無正當(dāng)理由逾期不予補(bǔ)正的,視為放棄申請,并將登記資料光盤按程序銷毀。 |
五、 受理審查決定
無實質(zhì)變化。從形式上來說,現(xiàn)在CDE只出具電子版的《受理通知書》《繳費(fèi)通知書》《檢驗通知書》《補(bǔ)正通知書》《不予受理通知書》,申請人在登記平臺上該登記號下面直接下載通知書PDF即可。
形式審查仍為5日。另外注意到的一點(diǎn)是,在受理流程圖中,增加了“推送《檢驗通知書》”。
|
受理審查決定 |
|
2020-07《化學(xué)藥品注冊受理審查指南(試行)》 |
2023-07《化學(xué)原料藥受理審查指南(試行)》 |
|
(一)受理 |
(一)受理 |
1.受理通知書:符合形式審查要求的,出具《受理通知書》一式兩份,一份給申請人,一份存入資料。2.繳費(fèi)通知書:需要繳費(fèi)。(二)補(bǔ)正申報資料不齊全或者不符合法定形式的,應(yīng)一次告知申請人需要補(bǔ)正的全部內(nèi)容,出具《補(bǔ)正通知書》。(三)不予受理不符合要求的,出具《不予受理通知書》,并說明理由。(四)受理流程圖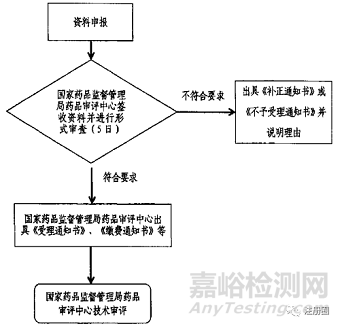 |
1.受理通知書:符合形式審查要求的,出具《受理通知書》(加蓋局行政許可受理專用章)。2.繳費(fèi)通知書:需要繳費(fèi)。(二)補(bǔ)正登記資料不齊全或者不符合法定形式的,應(yīng)一次告知登記人需要補(bǔ)正的全部內(nèi)容,出具《補(bǔ)正通知書》。(三)不予受理不符合要求的,出具《不予受理通知書》,并說明理由。(四)受理流程圖 |
總結(jié)
根據(jù)上述對比,原料藥登記人在今后登記原料藥時需要特別注意以下幾點(diǎn):
(1)資料形式及格式:藥品注冊申請電子文檔結(jié)構(gòu);光盤遞交;PDF文件,電子簽章。
(2)登記表:嚴(yán)格按填表說明填寫;注意核對碼一致性;未列入國家藥品標(biāo)準(zhǔn)或者藥品注冊標(biāo)準(zhǔn)的,應(yīng)當(dāng)提交通用名稱證明文件,或同時提出通用名稱核準(zhǔn)申請;登記人、生產(chǎn)企業(yè)、注冊代理機(jī)構(gòu)仍需簽字并加蓋公章。
(3)證明性文件:受理審查指南要求的都要提供,不要有任何遺漏;注意公證認(rèn)證要求;核對文件有效期,審評審批期間過期的需要及時更新。
(4)登記資料:光盤資料隨附《承諾書》;工藝驗證方案和報告必須有;穩(wěn)定性資料至少需要三個注冊批的6個月數(shù)據(jù);模塊一說明函內(nèi)容應(yīng)同時提交不適用的文檔清單及說明(如適用)。
(5)登記號管理:同一企業(yè)在同一生產(chǎn)場地生產(chǎn)的同一化學(xué)原料藥,生產(chǎn)工藝和質(zhì)量標(biāo)準(zhǔn)相同的,按照同一登記號登記;只要其中一個要素不一致,大概率就不能放到一個登記號下申報。
(6)審評期間變更:獲批后怎么變更都可以,但是審評期間不允許提交重大變更申請,不允許技術(shù)內(nèi)容變更,只有行政信息允許變更。
(7)通知書下載:提交登記資料5個工作日后,及時關(guān)注公示信息,下載《受理通知書》《繳費(fèi)通知書》《檢驗通知書》,及時繳費(fèi)、提交注冊檢驗樣品和資料;如收到《補(bǔ)正通知書》,在限期30日內(nèi)及時提交補(bǔ)正資料,補(bǔ)正時注意登記表是否需要更新,如需更新則需要重新簽字蓋章。
(8)需參考的法規(guī)指南:《藥品注冊管理辦法》、《國家藥監(jiān)局關(guān)于進(jìn)一步完善藥品關(guān)聯(lián)審評審批和監(jiān)管工作有關(guān)事宜的公告》、《化學(xué)藥品注冊分類及申報資料要求》《M4:人用藥物注冊申請通用技術(shù)文檔(CTD)》《國家藥監(jiān)局關(guān)于實施藥品注冊申請電子申報的公告》《關(guān)于藥品注冊申請電子申報有關(guān)要求的通知》《原料藥申報登記資料提交說明》。



