摘要
仿制藥的上市許可基于“平均”生物等效性(ABE)的證明,暴露量(全血、血清或血漿藥物濃度)幾何平均值的比率(仿制藥VS參比),90%置信區間(CI)的接受限度為0.8-1.25。然而,當在特定患者的治療中考慮到仿制藥和參比的互換性時,這種方法不能保證沒有治療影響,特別是對于治療指數較窄的藥物。本篇描述了ABE方法的基礎和局限性,以及監管機構提出的調整。對于高度可變的藥物,鑒于其較大的治療范圍,監管機構甚至允許擴大生物等效性的接受限度。對于治療指數較窄的藥物,監管機構對平均生物等效性方法進行了不同的修訂。歐洲藥品管理局只要求將ABE接受限值縮小至0.9-1.10范圍。美國食品藥品監督管理局(FDA)建議根據參比受試者內變異性縮小ABE的接受限值。FDA要求進行一項完全重復的交叉研究(四個周期),以便比較仿制藥和參比之間的受試者內變異性,并檢測受試者與制劑之間的相互作用。事實上,任何受試者內變異性的差異或受試者與制劑的相互作用,都是個體層面互換性的障礙。這些針對ABE的修正并沒有從根本上改變這樣一個事實,即個體暴露比率(仿制藥/參比)的變化比平均值的比率更大。由于實際原因和統計問題,無法進行真正的個體生物等效性研究,可以這樣建議,除了通常的平均生物等效性標準外,單個仿制藥/參比制劑暴露比率的95%置信區間限制也可以用于治療期間的互換性(至少對于治療指數較窄的藥物)。互換性接受的CI限值應按參比藥物的治療范圍進行縮放。監管機構可以根據生物等效性研究的真實數據集進行計算,以確定這些標準是否可以接受允許互換性。
1、引言
“平均生物等效性”(ABE)方法已用于仿制藥的上市許可多年。ABE是基于仿制藥和相應參比之間的暴露量幾何平均值(全血、血清或血漿藥物濃度)比率的置信區間(CI)確定。然而,監管機構并沒有在仿制藥的不同上市許可文件中指出,在對某一特定患者的治療過程中,用仿制藥替代(互換或轉換)與任何缺乏治療效果有關。
本文回顧了平均生物等效性方法的基礎和局限性,以及不同監管機構對受試者體內變異性大的藥物和治療指數窄的藥物所采取的調整措施。我們建議,為了允許在個體水平上的互換性,可以研究一個額外的標準,特別是對于治療指數較窄的藥物。除了平均值比率的CI以外,還可以考慮個體暴露比率(仿制藥/參比)的平均值CI。
關鍵點:
仿制藥的上市許可需要基于平均生物等效性方法(ABE)的生物等效性證明。
這并不能保證在給定患者的治療過程中,當參比藥物轉換為相應的仿制藥時,不會產生治療影響。
監管機構已建議對ABE進行調整,以適用于有較大受試者體內變異性的藥物和治療指數窄的藥物。
對于這種治療指數窄的藥物,建議在仿制藥與參比的個體暴露比的95%置信區間基礎上,提出一個允許互換性的額外標準。
2、平均生物等效性方法
2.1、“平均生物等效性”方法原理
用于仿制藥上市許可的生物等效性研究是基于一種多年來被不同監管機構接受的方法。它基于仿制藥活性物質與相應參比藥物活性物質的暴露方式(全血、血清或血漿藥物濃度)的比較。我們將簡化并考慮兩種產品最常見的情況,即口服給藥途徑和使用血漿藥物濃度暴露。研究的兩個暴露參數是血漿濃度-時間曲線下的面積(AUC)和峰值血漿濃度(Cmax)。生物等效性確認的一般原則是,如果仿制藥釋放的活性物質的平均暴露量與參比藥物的平均暴露量相差不超過20%,則認同為生物等效。使用這20%的標準是基于美國FDA的醫學專家的決定,他們發現,對于大多數藥物來說,血液中活性成分濃度的20%差異不會有臨床意義。
這意味著可以假設平均20%的差異在個體水平上不影響治療效果,或者活性物質在個體水平的效益/風險平衡。這一假設的藥理學基礎是,藥理作用(和治療效果)與任意活性物質的血液暴露動力學密切相關。事實上,藥物的效果取決于藥物作用部位的適當濃度,這個濃度是血液暴露量的函數,而血液暴露本身是物質吸收、分布、代謝、排泄的程度和速度的結果。
在ABE方法中,可以毫不費力的接受,在對某一特定病人開始新的治療時,提議用相應的仿制藥替代參比藥物。一般來說,這種替代會從治療的開始推斷到治療期間。然而,治療期間的互換性(或轉換),即在個體層面上沒有任何治療影響,不能由基于平均數的生物等效性證明來保證。
生物等效性研究通常在健康受試者身上進行,采用交叉設計,跨越兩個連續的時期,一個是仿制藥給藥,一個是參比給藥(圖1),有兩個給藥序列。如果活性物質的清除半衰期持續數天或數周,則可以采用平行組設計進行生物等效性研究。根據美國FDA或歐洲藥監局(EMA)的建議,通常是單劑量研究,根據藥代動力學的線性或非線性特征,選擇適當劑量。這種情況被認為是檢測不同制劑之間差異的最敏感情況。仿制藥的生物等效性研究只需要證明藥代動力學參數的生物等效性,而不用像生物仿制藥那樣需要證明額外的藥效學參數。
圖1 生物等效性研究:在10名健康受試者中,參比藥物和相應仿制藥中活性物質的血漿暴露比較。研究設計通常是交叉進行,有兩個暴露期(和兩個序列)。仿制藥和參比藥物之間的平均差異必須在參比藥物暴露量的±20%以內(見第2.1節)。連接仿制藥和參比之間暴露值的線的斜率在受試者之間沒有差異(沒有受試者與制劑之間的相互作用,仿制藥和參比的暴露在受試者內變異性也沒有差異)。AUC血漿濃度-時間曲線下的面積,Cmax峰值血漿濃度。
如果仿制藥和參比之間的暴露差異(AUC和Cmax)在-0.2×參比暴露量和+0.2×參比暴露量之間,則生物等效性成立,這也意味著仿制藥/參比暴露量的比率必須在0.8和1.20之間(表1,圖1)。
因此,必須對以下假設進行等效性檢驗:0.8<AUC仿制藥/AUC參比和0.8<AUC參比/AUC仿制藥;同樣的檢驗可用于Cmax。當同時考慮這兩個假設時,就會得出0.8<AUC仿制藥/AUC參比<1.25(因為如果0.8<AUC參比/AUC仿制藥,那么在數學上,AUC仿制藥/AUC參比<1/0.8=1.25;這就解釋了上限的1.25(而不是1.20),因為0.8是1.25的倒數。這就是Schuerman最早提出的用于證明生物等效性的所謂雙單側檢驗法。
根據監管機構所述的數學和統計學原因,仿制藥和參比之間暴露平均值的比較以及置信區間的計算是基于對數轉換后的暴露數據的方差分析(線性混合模型)。事實上,方差分析假設了一個加法模型,比較組之間的方差相等,以及正態分布的數據。數據的對數轉換符合這些要求。在這種條件下,初始非對數轉換數據的±20%區間(以0為中心)也可以變成以0為中心,因為0.8的對數值是-0.22,1.25的對數值等于+0.22。在對數尺度上,生物等效性的接受度遵循公式1:
ln(0.80)=-0.22<ln(仿制藥AUC)的平均值- ln(參比AUC)的平均值<ln 1.25=0.22 (1)
根據FDA的要求,生物等效性必須使用暴露數據的幾何平均值比率來證明。幾何平均值比率的90%置信區間(CI)必須落在0.8~1.25范圍內,總體α風險為5%(表1)。在對數尺度上的平均值與幾何平均值的比率(在線性非對數尺度上)之間的差異確實存在數學上的聯系:系列Xi對數值的算數平均值等于Xi幾何平均值的對數值。
表1 平均生物等效性的接受區間,基于兩種茶堿制劑在線性和對數尺度上數據平均值的差異和比率(改編自Rasheed和Siddiqui)
|
尺度
|
平均值的差異
|
平均值比率
|
|
線性
|
-0.20mr<(mg-mr)<0.20mp
|
0.80<(mg/mr)<1.20
|
|
對數尺度
|
-0.22314<(μg-μr)<0.22314
|
0.80<(μgeom g-μgeom r)<1.25
|
mr 參比藥物(線性尺度)的算數平均值(AUC或Cmax),mg 仿制藥(線性尺度)的算數平均值(AUC或Cmax);μr 參比藥物(對數尺度)的算數平均值(AUC或Cmax),μg 仿制藥(對數尺度)的算數平均值(AUC或Cmax);μgeom g 仿制藥的幾何平均值;μgeom r 為參比藥物的幾何平均值;AUC為血漿濃度-時間曲線下的面積;Cmax為藥物峰濃度。
log 0.8=-0.223124,log 1.25=+0.22314。log 0.8和log 1.25之間的區間在0附近是對稱的,而在1附近是不對稱區間0.8-1.25。
幾何平均值的比值是由對數值的算術平均值報告。事實上,系列Xi對數值的算數平均值等于Xi幾何平均值的對數值。
因此,生物等效性的評估是基于所考慮參數的總體幾何平均數(試驗/參比)比率的90%置信區間。該方法相當于在5%顯著水平下進行生物等效性零假設的兩次單側檢驗。
在雙期雙序列交叉設計的特殊情況下,當每個序列的個體數相同時(N/2),幾何平均數比率的對數的90%置信區間的界限計算如下(公式2):
XT-XR±t1-α×SE 標準誤差SE=S2 res(2/N)1/2 ………… (2)
S2 res是方查分析(以對數尺度進行分析)的殘余方差,N是交叉設計種納入的受試者數量,XT和XR分別是研究中觀測到的試驗藥物和參比藥物的幾何平均數的對數。
然后,在ABE中,根據公式2和3,幾何平均數比率(仿制藥/參比)的90%置信區間限度與血漿暴露的殘余方查(即受試者內變異性)的平方根成正比,與受試者人數的平方根成反比。
FDA公布了對1996年至2007年提交給監管機構的2070項ABE研究的統計分析:仿制藥與參比之間的暴露量平均差異為Cmax4.3%,AUC3.5%。在這些研究中,98%的仿制藥AUC與參比的差異平均不到10%。
2.2 平均生物等效性方法的局限性
2.2.1 受試者與制劑之間的相互作用以及仿制藥與參比之間的受試者內變異性差異
在交叉設計的生物等效性研究中,每個受試者有兩個暴露期(仿制藥/參比),方差統計分析假設仿制藥和參比之間的受試者內變異性方差相等,并且不考察受試者與制劑之間的任何相互作用。事實上,變異性的兩個來源,即仿制藥和參比之間的受試者內變異性方差以及受試者與制劑相互作用,都包含在交叉研究的方差分析的殘余方差中。當設計中只包含兩個暴露期,一個是仿制藥,另一個是參照物質時,它們無法被區分或單獨分析。
只有當交叉重復至少四個暴露期(至少兩個仿制藥和兩個參比),才能單獨分析仿制藥與參比之間的受試者內變異性差異以及受試者與制劑相互作用。
受試者與制劑相互作用(圖2)反映了一個結果,即在仿制藥和參比之間,不同受試者的暴露量可能有所不同。它可能會增加或減少。這可能是某些受試者亞組臨床特征的結果,這些臨床特征對仿制藥和參比之間活性物質的生物利用度有不同的影響。這在健康受試者中是相當理論化的,但在患者中是很有可能的。在這種情況下,如果仿制藥和參比之間的受試者內變異性差異保持不變,那么在這種相互作用產生了治療影響的情況下,必要時可以通過對受試者進行劑量適應性調整來解決這個問題。事實上,在重復給藥期間,這種受試者與制劑相互作用應保持相同的幅度。
圖2 生物等效性研究:受試者與制劑相互作用與六名受試者不同行為有關的圖示(仿制藥與參比之間的受試者內變異性沒有差異)。十個暴露期的假設結果。,每個個體在前五個時期接受參比藥物治療,后五個時期接受仿制藥治療。為了演示的目的,參比藥物和仿制藥的受試者內變異性很小(即,給定受試者的暴露量不會因為參比或仿制藥而發生重大變化)。受試者和制劑之間存在相互作用:當制劑發生變化時,受試者的暴露量會發生變化,而這些變化的幅度并不是所有患者都相同。AUC24h,從0到24小時的血漿濃度-時間曲線下的面積。
相反,仿制藥和參比之間受試者內變異性的差異(圖3)可能是仿制藥和參比之間不同崩解和溶解過程的結果,這可能影響活性物質的生物利用度。事實上,這是產品的質量問題,取決于其輔料,而仿制藥和參比之間輔料可能有所不同。這個問題導致了不同攝入量(在每日一次攝入量的情況下的日變異性)的活性物質利用率的受試者內變異性(仿制藥和參比之間),例如哌甲酯。在對同一受試者的治療過程中,這種現象會導致活性物質的生物利用度的升高或降低,如果變化幅度在特別敏感的患者中很重要,則可能對一些患者產生治療影響。重要的是,這種現象的隨機性使其無法通過劑量適應性進行任何的修正。
圖3生物等效性研究:仿制藥和參比之間受試者內變異性的圖示。假設6個人在前五個時期接受參比藥物治療,在后五個時期接受仿制藥治療。為了演示的目的,參比制劑的受試者內變異性較小。相比之下,仿制藥的受試者內變異性較高(即受試者的暴露量隨仿制藥制劑隨機變化)。受試者與制劑之間沒有相互作用,即對于每個受試者,兩種制劑的平均暴露量(在一段時間內)保持相同。在經典的兩期交叉試驗中(第5期和第6期),圖2和圖3中的情況下暴露量相同。因此,這種兩個周期的交叉試驗不能通過制劑相互作用將受試者內變異性的差異區分開來。它們都包含在方差分析(ANOVA)的殘差方差中。為了評估這種差異,至少需要在四個周期內重復交叉試驗(兩周期的參比和兩周期的試驗藥物)。AUC24h,從0到24小時的血漿濃度-時間曲線下的面積。
一般來說,交叉重復的生物等效性研究表明,仿制藥和參比之間的受試者內變異差異不大,與參比的受試者內變異相比,受試者與制劑之間的相互作用可以忽略不計。Concordet等人提出了受試者-制劑相互作用和受試者內變異性的差異,以此來解釋用新的Levothyrox®制劑替代舊制劑后觀察到的不良反應。
因此,假設受試者內變異性相同(仿制藥和參比之間),又缺乏對常見的兩期交叉設計的受試者與制劑相互作用的調查,使得ABE方法在開始治療時的替代是可以接受的,但它代表了反對在個體水平上的治療期間互換性保證的第一組論點,特別是當存在受試者內變異性的差異時,因為劑量適應性調整不能糾正這種差異,就像受試者與制劑相互作用一樣。
2.2.2 與仿制藥和參比之間血漿暴露量平均值附近的受試者之間分布有關的ABE的界限
ABE是基于仿制藥和參比暴露量的幾何平均值比率的90%CI區間。如第2.1段所述,CI值的寬度與納入受試者人數的平方根成反比。因此,納入受試者人數的增加會減少CI值的寬度。相反,仿制藥/參比單個暴露量比率的分布要大于其平均值比率計算的CI。這符合樣本分布和此類樣本平均值標準誤差之間的一般關系。
如圖4所示,這是輔料對活性物質的生物利用度的影響在受試者之間產生生物學差異的結果。
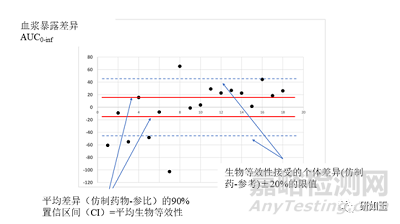
圖4兩種不同茶堿制劑之間暴露量(AUC)的個體差異(根據Rasheed和Siddiqui[10]報告的18名受試者的個體數據繪制)。平均生物等效性的界限是參比暴露量的±20%,此處為參比暴露量絕對值的±46.6(水平虛線)。18名受試者中有5名(28%)在該區間之外,盡管平均差異(試驗藥物-參比)的90%置信區間(CI)從-16.7至+15.6(水平連續線)證明了平均生物等效性;18名受試者中有12名(66%)在平均差異的90%置信區間(CI)之外。幾何平均值比率的90%CI=0.925-1.085(在0.8-1.25范圍內),點估計值=0.998。AUC0-inf,從0時到無限時間inf的血漿濃度-時間曲線下的面積。
因此,即使生物等效性可以用平均值的比率來證明,包括假設受試者內變異性沒有差異,也沒有任何受試者和制劑間相互作用,但在納入的樣本中受試者間的波動顯示,對一些患者來說,仿制藥和參比之間的暴露量差異會大于±20%,如圖4所示。個體數據來自Rasheed和Siddiqui發表的關于兩種茶堿制劑生物等效性研究的文獻。證明ABE的均值比90%CI范圍是0.92-1.8。然而,在納入的18名受試者中,有5人(27%)的個人仿制藥/參比暴露量的比率超出了這個范圍。
這意味著ABE方法不能保證個體水平的互換性,因為仿制藥和參比之間的暴露量差異可能大于可接受的±20%范圍,與此同時,平均值的差異確實在這個區間內。
這種情況下,治療效果取決于參比制劑的治療范圍。如果參比的治療范圍很大,并且遠大于20%的差異,那么在治療期間將仿制藥與參比制劑交換,不會產生任何治療影響。相反,如果治療范圍很窄,接近20%的接受范圍極限,個體波動可能導致療效和效益/風險平衡的治療變化。
為了解決ABE的這些不同的局限,監管機構和科學家們探索了其他生物等效性方法和針對ABE的調整,我們將在下一段進行介紹。
3、監管機構討論的解決ABE局限的方法
3.1個體生物等效性
在過去的20年里,統計學家和監管機構一直在爭論是否可以為生物等效性制定一個除ABE以外的方法,以便在個人層面上實現仿制藥和參比藥物之間的互換。“個體”生物等效性方法的想法已經出現,但結果相當混亂。事實上,為了在個體水平上建立生物等效性,理論上應在同一受試者身上多次重復給予仿制藥和參比藥物。這樣可以確定每個受試者的仿制藥和參比的暴露量平均值以及仿制藥和參比的受試者內變異性。我們可以直觀地理解,如果在重復給藥過程中觀察到仿制藥與參比暴露量之間的差異與不同參比給藥之間(以及不同批次參比之間)的暴露量相同(甚至更低),則可以認為個體生物等效性。換句話說,正如Chen等人[13]所述,這種個體生物等效性方法的目的是比較每個個體的仿制藥和參比的生物利用度差異(T-R)與參比制劑自身的生物利用度差異(R-R)。
這種在同一受試者身上重復給予仿制藥和參比藥物的個體生物等效性方法在實際中幾乎不可能執行。他們還假設受試者的臨床狀態隨著時間的推移完全保持穩定。這種個體生物等效性方法可能對患者更有價值,但在這種情況下,臨床狀態的穩定甚至比健康受試者更難獲得。
對于此類個體生物等效性研究,也提出了統計學問題,涉及周期數、接受限度、檢測能力等。
由于這兩個原因,這類個體生物等效性研究尚未納入常規實踐。
3.2 監管機構對受試者內變異性較大的藥物和治療指數較窄的藥物調整ABE的建議
3.2.1 受試者內變異性大的藥物
有些藥物所含的活性物質,在對某一患者重復給藥(相同劑量)期間,其生物利用度(和血液暴露量)存在較大的受試者內變異性。一般來說,這類物質表現為水溶解度低,親脂性低,生物利用度低,及肝臟首過效應。這些物質中的大多數屬于國際BCS分類(生物藥劑學分類)的第4類,低溶解性和低滲透性。
對于含有這種物質的藥物,鑒于受試者體內變異性較大,很難證明其生物等效性,與仿制藥的比較則更加困難。在這種情況下,生物等效性研究中所包含的受試者數量必須大大增加。
為解決受試者體內變異性大的藥物的這一問題,減少納入生物等效性研究的受試者數量,監管機構(如FDA和EMA)決定擴大生物等效性的接受限度,根據受試者內變異性進行調整。受試者內變異性是通過受試者內方差來衡量的,即交叉方差分析的殘差方差。變異系數(CV)來自殘差方差,其值接近于殘差方差的平方根。EMA對CV>30%,FDA對CV>20%提出了調整BE限度的建議。這種方法被稱為參比制劑校正平均生物等效性(RSABE)或擴展限度的平均生物等效性(ABEL)。FDA隨后建議擴大CV>25%的藥物的生物等效性接受限度,并將其應用于Cmax和AUC。EMA建議擴大CV>30%的藥物的限度,但僅用于Cmax。實際上,Cmax被認為比AUC更容易受到變異性的影響。此外,EMA建議對CV>50%的藥物,不再進一步擴大這些限度。
然而,這類受試者內變異性大的藥物通常有很大的治療范圍,否則它們就無法使用或者不能獲得上市許可。實際上,盡管血漿暴露量存在很大的波動(相同劑量連續兩次給藥之間超過20%),但其治療效果仍保持在同一水平。由于這個原因,仿制藥與參比制劑之間的互換性(已證明其平均生物等效性)并不令人擔憂,因為在較大的活性物質血液暴露量范圍內,療效和效益/風險比保持不變(圖5)。因為這些原因,擴展ABE的限值以允許互換性是可以接受的。
圖5 同一受試者服用治療范圍比較大的參比制劑(實線)和仿制藥(虛線)后,受試者內血漿暴露量的每日波動。即使仿制藥和參比之間存在一些受試者內變異性,活性物質的暴露量仍在治療范圍內。在這種情況下,互換不會產生任何治療影響。AUC24h,從0到24小時的血漿濃度-時間曲線下的面積。
3.2.2 治療指數窄的藥物
一些藥物及相應的活性物質具有狹窄的治療指數。然而,目前還沒有國際公認的此類物質的清單或定義的共同標準。對于這類藥物,不良反應可能發生在接近或略高于治療劑量時,具有突然的劑量-效應關系,通常需要進行治療藥物監測,即確定每個患者的血漿藥物濃度以進行劑量調整。這些藥物包括某些抗癲癇藥物、免疫抑制劑(他克莫司、環孢菌素、霉酚酸酯)、鋰制劑、地高辛、維生素K拮抗劑抗凝血劑和左旋甲狀腺素。
一般來說,治療指數較窄的藥物受試者內變異性較低(CV<30%)。如果不是這樣的話就不能使用。事實上,如果在同一患者中,治療指數窄的藥物從一次給藥到另一次給藥的暴露量變化很大,那么誘發藥物不良反應的概率或治療失敗的風險就很重要。
在大多數情況下,治療指數窄的藥物被用作慢性治療,在此期間,一種參比藥物可能被一種相應的仿制藥所取代,對這些藥物,提出了互換性的治療影響問題(圖6)。事實上,正是在這類藥物中,已經報道了療效的改變或效益/風險比的變化。抗癲癇藥物和甲狀腺素的情況尤其如此,其血漿暴露量±20%范圍內的平均生物等效性的常規接受標準受到了挑戰。
為了解決這個困難,監管機構提出了不同的策略來調整治療指數窄的藥物的平均生物等效性標準。EMA建議保持常見的兩期交叉設計,并將幾何平均值的仿制藥/參比的90%CI的接受限度定為0.9-1.11,而不是通常的0.80-1.25。
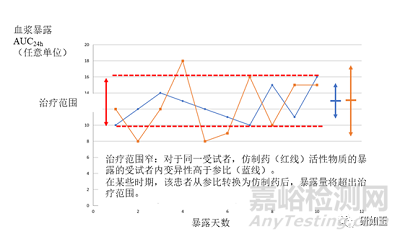
圖6 與圖5中的情況相同,但活性物質的治療指數較窄。同一受試者在試驗藥物和參比藥物之間的平均暴露量非常相似。然而,仿制藥(虛線)的受試者內變異性大于參比(實線),因此,在某些時候,仿制藥的暴露量可能超出治療范圍。在這種情況下,試驗藥物/參比藥物的互換性可能會對治療產生影響。AUC24h,從0到24小時的血漿濃度-時間曲線下的面積。
FDA建議將平均生物等效性的接受限度降低到活性物質的受試者內變異性區間(RSABE),最大限度降低到0.9-1.1。此外,FDA要求通過完成完全復制的交叉設計(每個受試者四個周期,兩個周期參比制劑,兩個周期仿制藥)來比較(F檢驗)仿制藥和參比之間的受試者內變異性。FDA接受仿制藥的受試者內變異性與參比之間沒有顯著性差異。該方法已針對華法林和左旋甲狀腺素進行了詳細介紹。復制交叉設計也可以檢測受試者與制劑的相互作用(參見第2.2.1段)。然而,正如第2.2.1段所述,受試者與制劑的相互作用可以通過仿制藥/參比轉換后的個體劑量調整來解決。相比之下,如果仿制藥的受試者內變異性高于參比,劑量調整將無法解決問題。這證明了FDA的立場,即要求對受試者內變異性進行比較,并拒絕受試者內變異性高于參比的仿制藥。
3.2.3 治療指數窄的藥物互換性對治療的影響
調整平均生物等效性的接受標準并不能解決個體水平上的互換性問題。事實上,正如前面在引言中提到的,監管機構并沒有聲稱平均生物等效性允許互換性;他們只是回避了這個問題。
Van Lancker等人對加巴噴丁的研究很好地說明了這個問題。對于這種抗癲癇藥物,在參比藥物和相應仿制藥之間切換后,已有關于治療影響和不良反應的報告。然后,他們進行了一項符合FDA要求的生物等效性研究,采用完全重復的交叉設計,將參比藥物與一種仿制藥(Sandoz仿制藥)進行比較。證實了平均生物等效性(幾何平均值的比率包含在0.8-1.25區間內),更有趣的是,他們還證明了受試者內變異性(仿制藥和參比之間)沒有差異,也沒有任何受試者-制劑相互作用。這項研究表明,即使在沒有受試者內變異性差異或受試者與制劑之間的相互作用的情況下,更換制劑時,也可能會產生治療影響(出現報告的不良反應)。
部分原因是個體暴露比率(仿制藥/參比)的一個樣本值的分布(95%置信區間)大于平均值比率的90%置信區間。如圖4所示。在Van Lancker發表的圖3中也說明了這一點,圖中顯示大多數的個體暴露比率(AUC)都在0.8-1.25的范圍之外。在Concordet等人[12]的研究中,比較了左旋甲狀腺素(l-甲狀腺素)的新舊制劑,他們還發現(通過計算),對于50%以上的患者,甲狀腺素(T4)暴露率的個體比值在縮小的0.9-1.11范圍之外,說明平均生物等效性并不能保證個體水平的生物等效。兩種左旋甲狀腺素制劑之間如此大的個體暴露比率范圍足以解釋在兩種制劑之間切換后報告的高不良反應率。
兩種左甲狀腺制劑之間的轉換是在非常大規模(超過200萬患者)和相當短的時間內(大約3-4個月)進行的,這解釋了可以檢測到這種藥物警戒信號。這種仿制藥/參比切換從未在如此大的規模和如此短的時間內執行。然后,在新舊制劑切換后,甲狀腺狀態出現治療失衡的患者比例非常小,這代表了相當重要的絕對病例數,可以被藥物警戒組織檢測到。事實上,在超過200萬名患者中,只有1.43%的患者報告了左旋甲狀腺激素轉換后的不良反應。
因此,有幾個假設可以解釋從一種參比藥物轉換為仿制藥后出現的治療影響和不良反應:兩種制劑之間暴露比率的個體分布大于平均值比率,仿制藥和參比之間所有受試者內變異性的差異,最后受試者與制劑的相互作用。就左旋甲狀腺素的具體情況而言,由于使用新制劑后不良反應逐漸消失,因此受試者內變異性有差異的假設不太可能。然而,在左旋甲狀腺素的生物等效性研究的兩期交叉試驗中,報告的CV值相對較大(23.7%),高于通常報告的左旋甲狀腺素的CV值,與新制劑的CV值較大是一致的,接近受試者內變異性高的藥物的30%界限。如前所述,這種情況不適合左旋甲狀腺素等治療指數窄的藥物。Concordet等人假設的受試者與制劑間的相互作用仍然是一種可能的解釋,但我們可以通過基于模擬的計算表明,在沒有這種相互作用的情況下,也可以獲得類似的生物等效性結果。左旋甲狀腺素案例中的另一個重要因素是,最初批準的舊制劑的片劑甲狀腺素含量較高(補償由于氧化降解而導致的甲狀腺素含量的逐漸下降),這在新制劑中不再允許。這種差異可能導致了血漿暴露量的差異,這些差異可能足以在非常敏感的患者中引起一些治療影響,盡管事實上已經證明了平均生物等效性。在左旋甲狀腺素這種情況下,防止這種治療影響的最佳方案是告知患者可能發生這種情況,并可能通過劑量調整來解決。
在治療指數窄的藥物的仿制藥/參比藥物轉換后報告出現治療不平衡的情況下,考慮到平均生物等效性方法在互換性應用方面的局限,一些監管機構建議不要轉換用這些治療指數較低的藥物治療的患者。一些監管機構建議不要給使用這些治療指數較窄的藥物治療的病人換藥。一些 "不轉換 "的藥物清單已經制定出來,特別是包括一些抗癲癇藥、一些免疫抑制劑和左旋甲狀腺素。在用這些藥物替代的情況下,如果治療影響是受試者-制劑相互作用的結果,可以根據治療反應進行適應性調整。如前所述,如果療效不平衡是由受試者內變異性造成的,那么任何劑量調整都不能解決這個問題(參見第2.2.2段)。然而,監管機構建議對治療指數較窄的藥物“不轉換”的立場,可能會使制藥公司不愿意用仿制藥的新配方來提高這種上市藥物的質量。
4. 除ABE標準之外的互換性補充標準的建議
為了解決仿制藥和參比制劑在個體水平上互換的可能性,我們建議監管機構在治療指數較窄的藥物的標準平均生物等效性接受標準中增加一些補充標準。此類標準應探討仿制藥和參比制劑之間暴露比率的個體值的分布。然后,除了平均值比率的CI外,還應考慮個體暴露比率(仿制藥/參比)的平均值的CI。可以建議,AUC和Cmax的單個暴露比值的95%置信區間應在先驗限值內,該先驗限值可以根據參比的治療范圍設置和縮放。例如,根據生物等效性的一般藥理學基礎,95%CI的限值可以是±20%,而對于治療指數較窄的藥物,平均生物等效性所要求的90%CI是±10%。我們建議,監管機構應根據生物等效性研究的真實數據集(他們擁有或可能要求制藥公司提供)進行計算和模擬,以檢測此類建議的可行性。
5.結論
在接受基于常規的平均生物等效性方法(ABE)的仿制藥上市授權后,當治療范圍遠大于平均生物等效性的一般接受限度時,仿制藥和參比制劑在治療期間的個體水平的互換性不會影響療效或效益/風險平衡。然而,對于治療指數較窄的藥物,當在個體層面上考慮仿制藥/參比互換性時,ABE方法不能保證不產生治療影響。由于這些原因,許多監管機構不建議對治療指數較窄的藥物進行仿制藥/參比轉換,除非可以根據治療反應進行劑量調整。除了常規的ABE標準外,監管機構可以對治療指數較窄的藥物進行補充標準檢測,以便在治療期間實現仿制藥/參比的互換。這種標準應基于仿制藥和參比制劑之間的單個暴露比的95%CI限值,并按參比制劑的治療范圍進行縮放。