您當(dāng)前的位置:檢測(cè)資訊 > 科研開發(fā)
嘉峪檢測(cè)網(wǎng) 2023-03-01 22:25
摘 要: 建立電感耦合等離子體質(zhì)譜法測(cè)定電泳涂料中15種金屬元素的分析方法。以微波灰化方式處理樣品,采用碰撞誘導(dǎo)解離(CID)-動(dòng)能歧視(KED)聯(lián)合模式消除質(zhì)譜干擾,實(shí)現(xiàn)高濃度元素和痕量元素的同時(shí)檢測(cè);采取氣體稀釋模式降低基體干擾并減少高濃度元素對(duì)質(zhì)量檢測(cè)器的損害。Na、Mg、Al、K、Ca、Fe和Zn質(zhì)量濃度在0.01~10.0 mg/L范圍內(nèi),Cu、Mn、Ni、Co、Cr、Cd、Ba和Pb的質(zhì)量濃度在1.0~1 000.0 g/L范圍內(nèi)線性關(guān)系良好,線性相關(guān)系數(shù)為0.999 1~0.999 9,方法檢出限為0.3~23 µg/kg。測(cè)定結(jié)果的相對(duì)標(biāo)準(zhǔn)偏差為0.6%~3.2%(n=6),樣品加標(biāo)回收率為91.5%~107.0%。該方法可用于汽車電泳涂料中金屬元素的檢測(cè)。
關(guān)鍵詞: 微波灰化; 電感耦合等離子體質(zhì)譜法; 電泳涂料; 金屬元素
電泳涂料作為汽車、機(jī)電等低污染的防腐涂料,主要成分是水溶性或水分散性離子型聚合物 [1],該類涂料已被多個(gè)國(guó)家廣泛采用。電泳涂料通過金屬離子導(dǎo)電在電解槽中實(shí)現(xiàn)涂裝,其中的金屬離子含量有嚴(yán)格的要求,雜質(zhì)離子含量過高會(huì)加速電解、電沉積[2],導(dǎo)致膜涂層針孔、陷穴、粗糙等弊病,嚴(yán)重時(shí)將造成凝聚和電泳特性失效。銅離子會(huì)使一些涂料產(chǎn)生過敏現(xiàn)象[3],國(guó)際上對(duì)環(huán)保型涂料中重金屬含量有嚴(yán)格的限制,因此金屬元素含量是電泳涂料工藝控制的重要環(huán)節(jié)。
我國(guó)涉及金屬元素檢測(cè)的涂料標(biāo)準(zhǔn)主要針對(duì)一些重金屬元素及有害物質(zhì)[4?6],而且需要先將涂料進(jìn)行涂膜后再消解處理。目前只規(guī)定了電泳涂料通用試驗(yàn)方法[7],缺乏電泳涂料中金屬元素的分析方法,相關(guān)文獻(xiàn)報(bào)道也較少,主要集中于重金屬檢測(cè)。陳勇等[8]以微波消解法分析了汽車涂料中重金屬;王友智等[9]以激光誘導(dǎo)擊穿光譜檢測(cè)了油漆中的重金屬含量;李凌偉等[10]以微波消解-ICP法測(cè)定了建筑涂料中重金屬元素。電泳涂料相關(guān)檢測(cè)方法報(bào)道少見。由于電泳涂料為水溶性,制膜困難,有機(jī)聚合物成分含量高,可溶性金屬萃取繁瑣,導(dǎo)致誤差較大。
干灰化法和微波消解法是常用的消解技術(shù),但存在消解時(shí)長(zhǎng)、樣品易損失、反應(yīng)劇烈、使用化學(xué)試劑及消解程度有限等缺陷。微波灰化法結(jié)合了高溫加熱和微波技術(shù),大幅縮短樣品制備時(shí)間,灰化效果更好,已應(yīng)用于石油、化工、食品檢測(cè)等領(lǐng)域[11?13]。涂料中金屬元素的限量范圍差異很大,采用電感耦合等離子體質(zhì)譜(ICP-MS)法[14?16]可以實(shí)現(xiàn)多元素同時(shí)分析且線性范圍較廣,能夠同時(shí)滿足不同濃度元素的檢測(cè)需求。
筆者采用微波灰化進(jìn)行前處理,采用氦氣碰撞誘導(dǎo)解離(CID)-動(dòng)能歧視(KED)模式和氣體稀釋優(yōu)化的ICP-MS法同時(shí)測(cè)定電泳涂料中的15種金屬元素,為電泳涂料檢測(cè)提供參考方法。
1 實(shí)驗(yàn)部分
1.1 質(zhì)譜條件
載氣流量:1.08 L/min;等離子氣流量:15 L/min;輔助氣流量:0.8 L/min;稀釋氣流量:0.2 L/min;氦氣流量:5.2 mL/min;RF功率:1 550 W;采樣深度:8 mm;霧化室溫度:2℃;蠕動(dòng)泵轉(zhuǎn)速:0.1 r/s;八級(jí)桿偏轉(zhuǎn)電壓:-18 V;QP偏轉(zhuǎn)電壓:-13 V;能量歧視電壓:5 V;四級(jí)桿真空度:1.32×10-5 Pa;氧化物比率:1.35%;雙電荷比率:0.82%。
1.2 樣品處理
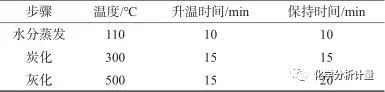
電泳涂料樣品超聲均質(zhì)后,稱取1 g,微波灰化程序見表1。
表1 微波灰化程序
樣品灰化后自動(dòng)冷卻至37 ℃以下,加入2%硝酸溶液5 mL,于180 ℃電熱板上加熱溶解,用2%硝酸轉(zhuǎn)移至200 mL石英容量瓶中,反復(fù)清洗坩堝內(nèi)壁,以2%硝酸溶液定容。靜置后取上清液上機(jī)檢測(cè)。同時(shí)做試劑空白。
2 結(jié)果與討論
2.1 樣品處理方法選擇
電泳涂料中含水量較高,傳統(tǒng)灰化法在水分蒸發(fā)過程中受熱不均,極易迸濺造成損失,因此需要先將其置于電熱板上蒸干,而蒸發(fā)和灰化時(shí)間過長(zhǎng),可達(dá)6~7 h;濕消解法和微波消解法取樣量受限,很難保證取樣的均勻性,且對(duì)有機(jī)物聚合物成分的消解較為困難;微波灰化法直接作用于樣品內(nèi)部水分子,加熱均勻,速度快,灰化徹底,因此選擇微波灰化法處理樣品。
2.2 樣品量選擇
若取樣量過小,則難以保證樣品均勻性;若取樣量過大,易導(dǎo)致樣品灰化不完全,同時(shí)產(chǎn)生大量氣體,容易飛濺。分別稱取0.5、1.0、1.5、2.0、3.0、5.0 g樣品進(jìn)行灰化試驗(yàn),結(jié)果表明當(dāng)樣品質(zhì)量小于2.0 g時(shí),灰化完成后樣品呈白色,能夠消解完全;但當(dāng)樣品質(zhì)量大于1.0 g時(shí)所需灰化時(shí)間較長(zhǎng),而當(dāng)樣品質(zhì)量小于0.5 g時(shí)測(cè)定結(jié)果的精密度較差,因此選擇稱樣質(zhì)量為1.0 g。
2.3 微波灰化程序的選擇
灰化溫度和升溫速率均會(huì)影響灰化效果。若溫度過低,則樣品灰化不完全;若溫度過高,則易造成元素?fù)p失。電泳涂料為液態(tài),升溫過快易造成樣品飛濺,升溫過慢則影響樣品處理效率。
采用階梯升溫保證樣品受熱均勻,即在常溫條件下放入馬弗爐中,加熱揮發(fā)水分,揮發(fā)完全后于低溫炭化,最后進(jìn)行高溫灰化。當(dāng)樣品在110 ℃保持10 min,水分可蒸發(fā)完全;若灰化溫度超過550 ℃,則Pb、Cd等元素回收率偏低。
考察炭化溫度分別為200、250、300 ℃,炭化時(shí)間分別為10、15、20 min,灰化溫度分別為450、500、550 ℃,灰化時(shí)間分別為15、20、25 min時(shí),電泳涂料樣品的灰化情況。以電泳涂料加標(biāo)樣品進(jìn)行正交試驗(yàn)L9(34),測(cè)定不同條件下待測(cè)溶液中各元素的濃度,并計(jì)算各元素回收率的平均值,考察上述因素的影響。微波灰化條件正交試驗(yàn)結(jié)果見表2,統(tǒng)計(jì)結(jié)果見表3。
表2 微波灰化條件正交試驗(yàn)L9(34)結(jié)果
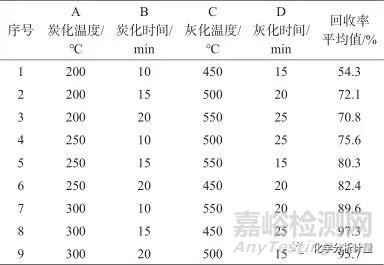
表3 微波灰化條件正交試驗(yàn)L9(34)統(tǒng)計(jì)結(jié)果

由表3可知,D2和D3即灰化時(shí)間20 min和25 min差別不大,選擇20 min。按照A3B2C2D2條件進(jìn)行微波灰化,樣品全部灰化為白色,測(cè)定待測(cè)溶液中各元素的回收率滿足要求,最終確定選擇微波灰化條件為炭化溫度300 ℃,炭化時(shí)間15 min,灰化溫度500 ℃,灰化時(shí)間20 min。
2.4 溶液體系和酸度選擇
ICP-MS分析受同量異位素干擾和分子離子干擾比較嚴(yán)重,特別是Ar、O、H、N、C等ICP中主要元素結(jié)合形成的分子和離子等;溶液中殘留的酸引入的基質(zhì)干擾也很高,因此應(yīng)盡量選擇不增加干擾離子的酸體系。使用鹽酸體系時(shí),易形成ClOH+、ClO+等干擾52Cr、55Mn等的測(cè)定,而硝酸引入的N、H、O等在基體中大量存在,因此選擇硝酸體系。
酸度引發(fā)基體效應(yīng),故盡量選擇低酸度。低于硝酸的質(zhì)量分?jǐn)?shù)1%時(shí)樣品中鹽類溶解不完全,故選擇硝酸溶液的質(zhì)量分?jǐn)?shù)為2%。
2.5 質(zhì)譜條件選擇
對(duì)多元素的同時(shí)測(cè)定,需要綜合考察質(zhì)譜工作參數(shù),分別選擇合適的等離子體模式、調(diào)諧模式和分析模式。
Na、Mg、Al、K、Ca、Fe和Zn元素含量(質(zhì)量分?jǐn)?shù))約為0.1%~10%,樣品溶液屬于高鹽基體,對(duì)等離子體會(huì)產(chǎn)生抑制效應(yīng),等離子體模式選擇高基體模式(HMI)即氣體稀釋方式,可以減少高鹽基質(zhì)鹽分引入、減少常量元素的質(zhì)譜響應(yīng),稀釋氣體比例選擇為1 : 8。
質(zhì)譜干擾主要為分子和多原子離子干擾,如39 K+、52Cr+、57Fe+、55Mn+、63Cu+分別受到38ArH+、40ArO+、40ArOH+、40Ar15N+、40Ar23Na+多原子干擾。采用八級(jí)桿反應(yīng)系統(tǒng)(ORS)的碰撞模式,同時(shí)使用氦氣碰撞誘導(dǎo)模式(CID)和動(dòng)能歧視模式(KED),利用He碰撞打斷分子離子鍵或者降低分子離子的動(dòng)能,利用碰撞池設(shè)置的勢(shì)能阱區(qū)分出低動(dòng)能的干擾離子消除上述分子離子干擾。待測(cè)離子因橫截面小、碰撞幾率小,故動(dòng)能損失較小而不受勢(shì)能阱影響,因此分析模式選擇He(CID-KED)模式。
霧化室中水汽引入40Ar16O+、40Ar1H+干擾56Fe+、41K+的測(cè)定,選擇霧化室溫度為2 ℃,可以有效降低干擾。
Cd元素激發(fā)能較高,選擇積分時(shí)間為1.0 s, Ca、Mg、Al和Fe元素含量高,選擇積分時(shí)間為0.10 s,其它元素積分時(shí)間為0.30 s。
2.6 同位素質(zhì)量數(shù)和內(nèi)標(biāo)元素選擇
電泳涂料中的Na、Mg、Al、K、Ca、Fe和Zn含量相對(duì)較高,考慮選擇同位素豐度相對(duì)較低的質(zhì)量數(shù),以擴(kuò)大線性范圍、減少稀釋概率。Na和Al沒有同位素,54Fe和54Cr相互干擾,40Ca和56Fe分別受到40Ar、40ArO+干擾,因此選擇43Ca和57Fe作為分析質(zhì)量數(shù)。Pb元素多同位素同時(shí)存在,同時(shí)監(jiān)測(cè)206Pb、207Pb和208Pb,使用干擾方程計(jì)算208Pb作為鉛元素含量。
質(zhì)量數(shù)相近的元素物理性質(zhì)相似,能更好反應(yīng)待測(cè)元素受到干擾的情況,45Sc、72Ge、115In、209Bi在樣品中幾乎不存在,分別作為低、中、高質(zhì)量數(shù)的內(nèi)標(biāo)元素用于校正基體干擾。
3 結(jié)論
建立了微波灰化-ICP-MS法測(cè)定電泳涂料中Na、Mg、Al、K、Ca、Fe、Zn、 Cu、Mn、Ni、Co、Cr、Cd、Ba和Pb 15種元素含量的分析方法。
采用正交試驗(yàn),選擇微波灰化條件進(jìn)行樣品前處理,再采用氦氣碰撞誘導(dǎo)解離-動(dòng)能歧視模式和氣體稀釋消除質(zhì)譜干擾和基體干擾,減少稀釋倍數(shù)。方法學(xué)考證結(jié)果表明,該方法實(shí)現(xiàn)了多種元素的同時(shí)檢測(cè),操作簡(jiǎn)便,線性范圍較寬,測(cè)定結(jié)果準(zhǔn)確、可靠。樣品在80 min內(nèi)灰化完全,該方法適合應(yīng)用于批量檢測(cè)。
該方法可為電泳涂料工藝和質(zhì)量控制的研究提供參考,但微波灰化法加熱溫度較高,分析低溫元素尚有一定的局限性,有待于尋找更方便、快捷、全面的分析方法同時(shí)滿足檢測(cè)需求。

來源:化學(xué)分析計(jì)量


