摘 要 / Abstract
目的:通過介紹510(k)(上市前通知)第三方審查計劃及優(yōu)化措施,為我國醫(yī)療器械審評制度創(chuàng)新提供相關(guān)參考。方法:查閱FDA官網(wǎng)相關(guān)指南和文件,對第三方審查機(jī)構(gòu)認(rèn)可資質(zhì)、審評程序、監(jiān)督機(jī)制進(jìn)行介紹。結(jié)果:第三方審查計劃的實(shí)施對彌補(bǔ)監(jiān)管資源、緩解審評壓力有所幫助。結(jié)論:建議我國可以嘗試引入第三方機(jī)構(gòu)協(xié)助承擔(dān)部分醫(yī)療器械上市前技術(shù)審評任務(wù),減輕監(jiān)管部門工作負(fù)擔(dān),提高審評效率和質(zhì)量。同時需要規(guī)范審評流程、完善相關(guān)配套法律法規(guī)、重視溝通交流機(jī)制的建立及加強(qiáng)對第三方機(jī)構(gòu)的監(jiān)督。
Objective: To provide insights for innovating the medical device review system in China by introducing the 510(k) (Premarket Notification) Third Party Review Program of the US FDA. Method: By studying relevant guidelines and documents on the FDA website, an introduction is given on the accreditation qualifications of third-party review organizations, review procedures and the oversight mechanism. Results: Implementation of the third party review program is helpful to supplement regulatory resources and ease review pressure. Conclusion: Third party organizations may be introduced in China to assist in the pre-market technical review tasks of some medical devices, with the aim of reducing regulatory burden and improving review efficiency and quality. At the same time,efforts should be made to standardize the review process,improve relevant laws and regulations, facilitate communication mechanisms, and strengthen supervision of third-party organizations.
關(guān) 鍵 詞 / Key words
510(k);第三方審查計劃;醫(yī)療器械審評制度
510(k); third party review program; medical device review system
1、引 言
與藥品相比,醫(yī)療器械門類更多,學(xué)科跨度更大。兩者都是關(guān)系國計民生和人民生命健康的特殊商品,必須嚴(yán)格做到安全、有效與質(zhì)量穩(wěn)定。產(chǎn)品的質(zhì)量體系及其運(yùn)作水平是影響產(chǎn)品質(zhì)量的重要因素。隨著科學(xué)技術(shù)的迅猛發(fā)展,尤其是計算機(jī)和人工智能的快速發(fā)展,醫(yī)療器械產(chǎn)業(yè)隨之高速發(fā)展,其產(chǎn)品與質(zhì)量體系越來越復(fù)雜,藥品監(jiān)管部門對紛繁復(fù)雜的醫(yī)療器械產(chǎn)品與產(chǎn)品質(zhì)量管控體系進(jìn)行審計和檢查的工作量和難度與日俱增。國務(wù)院于2015年發(fā)布的《關(guān)于改革藥品醫(yī)療器械審評審批制度的意見》提倡:“將食品藥品監(jiān)管總局列為政府購買服務(wù)的試點(diǎn)單位,通過政府購買服務(wù)委托符合條件的審評機(jī)構(gòu)、高校和科研機(jī)構(gòu)參與醫(yī)療器械和仿制藥技術(shù)審評、臨床試驗(yàn)審評、藥物安全性評價等技術(shù)性審評工作。”[1]2017年10月,中共中央辦公廳、國務(wù)院辦公廳印發(fā)的《關(guān)于深化審評審批制度改革鼓勵藥品醫(yī)療器械創(chuàng)新的意見》提倡:“將藥品醫(yī)療器械審評納入政府購買服務(wù)范圍,提供規(guī)范高效審評服務(wù)。”[2]在美國、歐盟等發(fā)達(dá)國家和地區(qū),第三方機(jī)構(gòu)已廣泛參與醫(yī)療器械產(chǎn)品的審評和質(zhì)量管理體系核查等活動[3],社會力量得到充分利用,監(jiān)管效率也逐步提升。通過研究和學(xué)習(xí)美國關(guān)于第三方機(jī)構(gòu)參與醫(yī)療器械產(chǎn)品的審評和質(zhì)量管理體系核查的具體做法,對于我國深化醫(yī)療器械行業(yè)發(fā)展具有重要意義。
2、510(k)第三方審查計劃概述
510(k),又稱為上市前通知,是產(chǎn)品進(jìn)入美國市場的一種上市途徑。任何國家的設(shè)備銷售方計劃在美國開展銷售都必須先向美國食品藥品監(jiān)督管理局(FDA)提交上市前文件,證明自己即將上市的設(shè)備與市面上已經(jīng)合法銷售的設(shè)備具有同等安全性和有效性[4],且只有通過了510(k)審查,設(shè)備才可正常上市銷售。《聯(lián)邦食品藥品和化妝品法案》(Federal Food,Drug and Cosmetic Act,F(xiàn)D&C Act)第523條規(guī)定,允許經(jīng)過認(rèn)可的第三方機(jī)構(gòu)審查某些中低風(fēng)險的醫(yī)療器械。FDA并于2020年3月12日頒布了《針對行業(yè)、食品和藥品管理局工作人員和第三方審查組織的第三方審查方案指南》[510(k) Third Party Review Program Guidance for Industry Food and Drug Administration Staff and Third Party Review Organizations][5],該指南對第三方機(jī)構(gòu)的準(zhǔn)入資格、審評的設(shè)備范圍及審查的具體程序等都進(jìn)行了嚴(yán)格規(guī)定。美國實(shí)施510(k)第三方審查計劃,旨在幫助FDA更快地做出審評決策,為醫(yī)療器械制造商提供自愿的替代審查程序,使FDA可將資源和精力主要集中在高風(fēng)險設(shè)備的審查上,但同時掌握第三方機(jī)構(gòu)所審查的低風(fēng)險設(shè)備的最終決定權(quán)。
2.1 第三方機(jī)構(gòu)認(rèn)可要求
第三方機(jī)構(gòu)的認(rèn)可包括首次認(rèn)可和繼續(xù)認(rèn)可。FDA法規(guī)文件主要從政務(wù)信息、防止利益沖突、人員資質(zhì)及保證聲明四個方面作為首次認(rèn)可的考慮因素。第三方機(jī)構(gòu)需要向FDA提供基礎(chǔ)信息,同時保證參與510(k)審評的人員與設(shè)備銷售方之間無利益關(guān)聯(lián),防止任何可能影響審查過程的利益沖突出現(xiàn),并簽署保證聲明。
其中FDA對于審評人員的要求采用的是國際醫(yī)療器械監(jiān)管者論壇(International Medical Device Regulators Forum,IMDRF)2017年發(fā)布的《監(jiān)管審查員的能力、培訓(xùn)和行為要求》(Competence,Training,and Conduct Requirements for Regulatory Reviewers)[6](文件編號170316),具體要求詳見表1。
2.2 第三方機(jī)構(gòu)審評程序
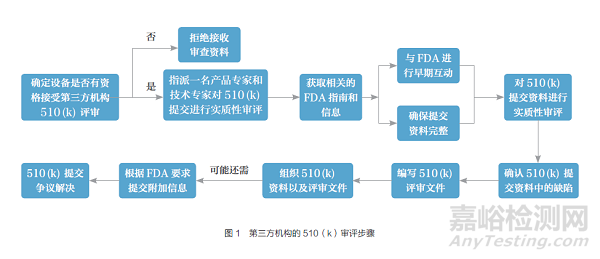
第三方機(jī)構(gòu)具體審評程序主要分為7步,具體流程如圖1所示。
2.2.1 確定第三方機(jī)構(gòu)審評范圍
FDA根據(jù)醫(yī)療器械風(fēng)險高低、特殊控制或一般控制是否足以對醫(yī)療器械的安全有效性提供合理的保證等因素將醫(yī)療器械劃分為Ⅰ類、Ⅱ類和Ⅲ類,只有風(fēng)險較低的I類及部分Ⅱ類醫(yī)療器械能夠參與第三方審評計劃。第三方機(jī)構(gòu)在開展審評前需要根據(jù)產(chǎn)品代碼分類數(shù)據(jù)庫或FDA第三方審評公共網(wǎng)站進(jìn)行設(shè)備審評資格的查詢,同時還需判斷自己是否具有審評該設(shè)備類型的專業(yè)知識和能力。只有兩個條件同時滿足,才能開展審評工作。FDA審查部門表明,第三方機(jī)構(gòu)提交的低風(fēng)險設(shè)備的510(k)審評意見不需要復(fù)審,而復(fù)雜設(shè)備的510(k)重新審評可能性較大。為了減少復(fù)審次數(shù),提高審評效率,F(xiàn)DA計劃調(diào)整審評名單,將需要FDA額外審評的醫(yī)療器械從名單中移除。
2.2.2 確保提交資料的完整性
第三方機(jī)構(gòu)根據(jù)FDA相關(guān)法律法規(guī)要求對申請者提交的510(k)進(jìn)行驗(yàn)收評審,查看資料的完整性。如果資料不完整,第三方機(jī)構(gòu)應(yīng)拒絕開展審評。
2.2.3 實(shí)質(zhì)性審評
實(shí)質(zhì)性審評的重點(diǎn)是確定申請上市的醫(yī)療器械是否與市面上已經(jīng)合法銷售的醫(yī)療器械有著相同的安全性與有效性。第三方機(jī)構(gòu)需要大量查閱FDA的相關(guān)數(shù)據(jù)庫,獲取有關(guān)的指導(dǎo)文件及信息,如上市前評審信息、上市后召回、醫(yī)療器械報告,以及申請者提供的書面反饋、會議紀(jì)要及其他數(shù)據(jù)。FDA 也會為第三方機(jī)構(gòu)提供培訓(xùn)和專家咨詢等幫助其完成審評。
2.2.4 早期互動
510(k)審評期間,第三方機(jī)構(gòu)會與FDA進(jìn)行早期互動,尤其是在審評從未接觸過的醫(yī)療器械類型之前,這有助于第三方機(jī)構(gòu)了解FDA對該設(shè)備類型最新的相關(guān)指導(dǎo)原則、標(biāo)準(zhǔn)和其他考慮事項(xiàng),識別相關(guān)問題,確保審評內(nèi)容和現(xiàn)行審評標(biāo)準(zhǔn)一致。溝通方式通常采用電話或郵件。
2.2.5 缺陷處理
第三方機(jī)構(gòu)在審評過程中發(fā)現(xiàn)任何缺陷應(yīng)立即聯(lián)系510(k)提交人。第三方機(jī)構(gòu)可以采用電話、傳真、電子郵件或信函等方式來解決問題,避免僅通過電話交換實(shí)質(zhì)性數(shù)據(jù)和信息,需要有書面請求和答復(fù),同時必須進(jìn)行保密和記錄。如果510(k)申請者為了解決缺陷對提交資料進(jìn)行了任何修改,第三方機(jī)構(gòu)需要進(jìn)行實(shí)時記錄,并要求510(k)申請者及時提供510(k)的最新版本。
2.2.6 編寫審評文件
第三方機(jī)構(gòu)在提出審評建議時需要編寫審評備忘錄,提供定制的審評備忘錄模板,說明做出最終建議的理由和步驟,并且清楚地記錄所有缺陷、對缺陷的響應(yīng)及對審評的響應(yīng)。FDA為第三方提供了定制的審評備忘錄模板。審評備忘錄是FDA對第三方機(jī)構(gòu)審評工作情況進(jìn)行了解的唯一途徑,清晰簡明的文件可減少FDA對510(k)的審評次數(shù),提高審評效率。
2.2.7 提交510(k)資料和審評意見
最終審評人員在完成510(k)審評意見的審核后,應(yīng)向FDA文件控制中心提交兩個獨(dú)立的文件,即申請者提交的510(k)提交文件(最新版本)和機(jī)構(gòu)編寫的審評文件,其中審評文件一般包括附有最終審查員簽字的相關(guān)基礎(chǔ)信息表、申請者授權(quán)給第三方機(jī)構(gòu)的510(k)、保證性聲明、文檔目錄、早期交互信息、審評備忘錄等。
FDA只有在必要文件齊全后,才會對第三方機(jī)構(gòu)的審評備忘錄進(jìn)行審評,同時可能也會重新審評申請者提交的510(k)。如果FDA認(rèn)為需要額外的信息,將發(fā)送電子郵件通知第三方機(jī)構(gòu)暫停工作,并要求其提供附加信息。
2.3 監(jiān)管機(jī)構(gòu)對第三方機(jī)構(gòu)的監(jiān)督
第三方機(jī)構(gòu)在通過首次認(rèn)可之后還需要接受FDA的繼續(xù)認(rèn)可,繼續(xù)認(rèn)可由醫(yī)療器械和放射健康中心(Center for Devices and Radiological Health,CDRH)的投訴監(jiān)督辦公室(OC)依法定期開展(至少每三年一次),檢驗(yàn)第三方機(jī)構(gòu)是否按照法規(guī)的要求進(jìn)行相關(guān)審核工作并記錄過程,并且依然滿足認(rèn)可標(biāo)準(zhǔn)。繼續(xù)認(rèn)可因素包括第三方機(jī)構(gòu)過去上市前的評估表現(xiàn)和正在進(jìn)行的定期評估表現(xiàn)。良好的歷史記錄和現(xiàn)行審查業(yè)績記錄可表明第三方機(jī)構(gòu)能夠出具與FDA等效的建議和評審。FDA通過使用公開可用的指標(biāo)(第三方審評組織績效報告)來監(jiān)控第三方機(jī)構(gòu)和整個審評計劃的實(shí)施,定期進(jìn)行持續(xù)改進(jìn)分析,以確定計劃中可能需要改進(jìn)的部分,并進(jìn)行相應(yīng)地調(diào)整[7]。
當(dāng)有足夠的證據(jù)表明,第三方審核機(jī)構(gòu)與申報者之間存在著利益關(guān)系,或未按照標(biāo)準(zhǔn)和法規(guī)的要求履行程序,對公眾健康造成威脅時,F(xiàn)DA可以在提供非正式聽證會的機(jī)會之后采取措施暫停或撤銷第三方審評機(jī)構(gòu)資格。
3、對我國醫(yī)療器械審評制度的思考
3.1 引入第三方審核的優(yōu)勢和局限性
3.1.1 優(yōu)勢
雖然我國形成了以國家藥品監(jiān)督管理局醫(yī)療器械技術(shù)審評中心、省級醫(yī)療器械審評中心及2個醫(yī)療器械技術(shù)審評檢查分中心(國家藥品監(jiān)督管理局醫(yī)療器械技術(shù)審評檢查長三角分中心、國家藥品監(jiān)督管理局醫(yī)療器械技術(shù)審評檢查大灣區(qū)分中心)構(gòu)成的醫(yī)療器械技術(shù)審評體系,并通過招聘、編制、借調(diào)等方式不斷引入人才、擴(kuò)充技術(shù)審評力量,但是審評資源相對缺乏的現(xiàn)狀仍然存在,審評人員的數(shù)量與高質(zhì)量完成審評任務(wù)的需求還存在差距。引入第三方機(jī)構(gòu)參與審評工作,可以有效緩解監(jiān)管部門審評資源不足的情況。以美國統(tǒng)計的平均值為例,監(jiān)管部門對風(fēng)險較低的簡單醫(yī)療器械進(jìn)行單獨(dú)審評需要105天,在第三方機(jī)構(gòu)輔助審評需要74天,縮短了31天;同樣,對風(fēng)險較高的復(fù)雜醫(yī)療器械單獨(dú)審評需要156天,在第三方機(jī)構(gòu)輔助為83天,縮短了73天[8],審評效率顯著提升。
3.1.2 局限性
雖然引入第三方機(jī)構(gòu)力量可以獲益,但是監(jiān)管部門若無法對第三方機(jī)構(gòu)進(jìn)行有效地監(jiān)管,則該措施亦存在一定的風(fēng)險。一方面由于第三方機(jī)構(gòu)專業(yè)技術(shù)能力不足或者不同審評員對法規(guī)要求的理解不同,對產(chǎn)品獲益-風(fēng)險評估的標(biāo)準(zhǔn)不同,主觀性較強(qiáng),容易導(dǎo)致審評結(jié)果出現(xiàn)紕漏,使得安全有效性存在較大風(fēng)險的問題產(chǎn)品上市,造成安全事故發(fā)生的風(fēng)險。例如2011年法國某公司的植入性產(chǎn)品通過第三方機(jī)構(gòu)完成CE認(rèn)證后上市,引發(fā)了多例不良事件,之后法國的衛(wèi)生部門重新評估后認(rèn)為該產(chǎn)品對人體存在不可接受的風(fēng)險,不宜上市。另一方面是信任風(fēng)險,第三方機(jī)構(gòu)作為商業(yè)組織,存在一定的逐利性,可能難以避免地會出現(xiàn)權(quán)力尋租的現(xiàn)象,一旦發(fā)生,將給人民群眾的安全帶來巨大風(fēng)險。因此,如何對第三方機(jī)構(gòu)進(jìn)行監(jiān)督和管理是該計劃實(shí)施過程的重點(diǎn)難點(diǎn)之一。
3.2 其他國家或地區(qū)引入第三方機(jī)構(gòu)情況
3.2.1 歐盟
歐盟的第三方認(rèn)證機(jī)構(gòu)稱為公告機(jī)構(gòu),是由歐盟各成員國指定并通過所在成員國相關(guān)認(rèn)證機(jī)構(gòu)的認(rèn)可后成立的。歐盟委員會將評估合格的公告機(jī)構(gòu)名單公布于NANDO(New Approach Notified and Designated Organisations)網(wǎng)站[9],設(shè)備制造商可通過瀏覽該網(wǎng)站,自行選擇公告機(jī)構(gòu)進(jìn)行醫(yī)療器械合格評定。合格評定結(jié)束后,公告機(jī)構(gòu)會為其頒發(fā)CE證書,該醫(yī)療器械才可在歐盟市場中流通和使用。與美國不同的是,歐盟公告機(jī)構(gòu)不僅擁有產(chǎn)品技術(shù)資料審評權(quán),還能直接決定產(chǎn)品能否上市并負(fù)責(zé)產(chǎn)品的上市后監(jiān)管。歐盟目前采用最新版醫(yī)療器械法規(guī)[Regulation(EU)2017/745,簡稱MDR]對公告機(jī)構(gòu)進(jìn)行嚴(yán)格管理。
3.2.2 加拿大
加拿大將醫(yī)療器械分成四類,其中I類風(fēng)險最低,IV風(fēng)險最高。醫(yī)療器械上市前審評實(shí)行的是注冊資料審查和質(zhì)量體系審查相結(jié)合的方式[10],與我國相類似,但其質(zhì)量體系核查是由加拿大醫(yī)療器械符合性評估系統(tǒng)(CMDCAS)認(rèn)可的第三方認(rèn)證機(jī)構(gòu)(Canadian Medical Devices Conformity,CMDCAS)承擔(dān),指導(dǎo)文件為GD210∶ISO 13485∶2003質(zhì)量管理體系審核[11]。加拿大衛(wèi)生部僅接受第三方審核機(jī)構(gòu)頒發(fā)的質(zhì)量體系證書。Ⅱ、Ⅲ、Ⅳ類醫(yī)療器械在申請注冊時,需要提交第三方機(jī)構(gòu)出具的醫(yī)療器械質(zhì)量管理體系認(rèn)證證書。
3.3 啟示
3.3.1 審評機(jī)制探索,開展試點(diǎn)工作
鑒于美國和其他發(fā)達(dá)國家和地區(qū)第三方審核計劃所取得的良好效果與經(jīng)驗(yàn),筆者建議我國可嘗試開展第三方機(jī)構(gòu)輔助審核試點(diǎn)工作。美國的第三方審核計劃遵循自愿原則,由申請人將費(fèi)用直接支付第三方機(jī)構(gòu),F(xiàn)DA不收取任何費(fèi)用,使得第三方機(jī)構(gòu)可以獲得適當(dāng)利潤,也可縮短設(shè)備的審核時間,加快上市速度。國外現(xiàn)在已經(jīng)有許多較為成熟的第三方審評機(jī)構(gòu),如英國標(biāo)準(zhǔn)協(xié)會(British Standards Institution)等。我國可以先與跨國認(rèn)證機(jī)構(gòu)進(jìn)行合作,實(shí)現(xiàn)對人才、先進(jìn)技術(shù)、理念和審評標(biāo)準(zhǔn)的引進(jìn)[12],選取少數(shù)地區(qū)開展試點(diǎn)工作,并確定審評目錄,可將第一類備案產(chǎn)品和少部分風(fēng)險較低的第二類產(chǎn)品交與第三方審評,但第三方只負(fù)責(zé)給出其審評建議,最終審批權(quán)還是掌握在審評部門。根據(jù)試點(diǎn)效果反饋,如反饋情況良好,可以適當(dāng)擴(kuò)大試點(diǎn)范圍,同時出臺鼓勵政策推動國內(nèi)第三方機(jī)構(gòu)的發(fā)展,如稅收優(yōu)惠、資金補(bǔ)貼等。醫(yī)療器械具有高風(fēng)險性,第三方機(jī)構(gòu)的準(zhǔn)入門檻需設(shè)定較高標(biāo)準(zhǔn),可以從審評人員資質(zhì)、操作要求、工作的公正性和保密性、防止利益沖突或聯(lián)結(jié)等方面設(shè)定第三方機(jī)構(gòu)的認(rèn)可資質(zhì)。
3.3.2 出臺配套法規(guī),肯定合法地位
利用第三方機(jī)構(gòu)的力量輔助監(jiān)管部門事務(wù)在許多發(fā)達(dá)國家和地區(qū)中屢見不鮮,取得的成效有目共睹。但目前我國醫(yī)療器械法規(guī)體系尚未明確第三方機(jī)構(gòu)參與監(jiān)管部門輔助工作的合法地位,這對第三方機(jī)構(gòu)在我國的發(fā)展產(chǎn)生了一定的阻礙。建議我國出臺對第三方機(jī)構(gòu)進(jìn)行規(guī)范管理的配套法律法規(guī),完善現(xiàn)有法律體系,一方面確定第三方機(jī)構(gòu)合法地位、權(quán)利、義務(wù)及出現(xiàn)問題時各方應(yīng)承擔(dān)的責(zé)任;另一方面對第三方的行為進(jìn)行必要約束和規(guī)范,如制訂泄露用戶信息的懲罰措施等,增強(qiáng)審評的合法性,使醫(yī)療器械生產(chǎn)企業(yè)可以安心、主動選擇第三方機(jī)構(gòu)進(jìn)行技術(shù)審評,提高醫(yī)療器械上市速度,推進(jìn)醫(yī)療器械行業(yè)快速發(fā)展。
3.3.3 加強(qiáng)事前交流,完善溝通機(jī)制
FDA十分重視溝通反饋,鼓勵制造商與CDRH在醫(yī)療器械審評的各個環(huán)節(jié)開展溝通交流,并將申請人對FDA提出的各種溝通反饋請求統(tǒng)稱為Q-Submissions。Q-Submissions包括正式提交申報資料前的資料預(yù)提交(pre-sub)、信息報告會、研究風(fēng)險確定會議、早期正式合作會議、審評過程中的資料提交問題分析會和上市前審批(PMA)100天會議[13]。第三方機(jī)構(gòu)與FDA 之間也采取了郵件、電話等方式進(jìn)行早期互動。良好的溝通可以大幅提高工作效率,目前的立卷審查制度、特殊產(chǎn)品溝通機(jī)制是我國進(jìn)行審評前移非常有益的探索,建議我國進(jìn)一步加快和完善溝通交流機(jī)制,如建立第三方與省級審評機(jī)構(gòu)、國家安全技術(shù)部門專門的溝通渠道,并增設(shè)咨詢平臺,創(chuàng)新溝通方式,除電話和郵件外,可采取定期開座談會、視頻會,創(chuàng)建交流群等方式收集反饋意見,加強(qiáng)注冊全過程的交流、答疑及探討,提高工作效率和質(zhì)量。
3.3.4 規(guī)范審評流程,統(tǒng)一審評尺度
審評尺度不一致是我國醫(yī)療器械審評過程存在的主要問題之一[14]。醫(yī)療器械涉及科目廣泛,不同專業(yè)的審評人員對審核產(chǎn)品的理解不同,這可能會造成對同一產(chǎn)品的審核側(cè)重點(diǎn)和補(bǔ)正意見存在不同,使得企業(yè)對材料反復(fù)修改,延長審評時間。為了確保第三方機(jī)構(gòu)審評工作的順利實(shí)施及審評結(jié)果的可靠性,建議相關(guān)部門對第三方機(jī)構(gòu)審評流程和審評標(biāo)準(zhǔn)進(jìn)行統(tǒng)一規(guī)定,可以參考其他國家和地區(qū)規(guī)定的第三方審評流程,編寫審評模板,并對審評人員進(jìn)行統(tǒng)一集中培訓(xùn),提高審評的正確性,減少重新審查次數(shù)。
3.3.5 嚴(yán)格監(jiān)督機(jī)制,完善監(jiān)管體系
第三方機(jī)構(gòu)作為獨(dú)立的法人組織,可能存在一定的逐利性。為了保證審評過程的公正性,第三方機(jī)構(gòu)除了保證自身自律,還必須接受監(jiān)管部門、企業(yè)、同行及社會的監(jiān)督。建議監(jiān)管部門對第三方機(jī)構(gòu)建立績效考評機(jī)制和動態(tài)退出機(jī)制,可以根據(jù)第三方機(jī)構(gòu)績效報告、投訴舉報及不良事故發(fā)生率等情況對其進(jìn)行考察和評級,并將考評結(jié)果及時向大眾公布。對于不合格的第三方機(jī)構(gòu),監(jiān)管部門可根據(jù)結(jié)果的嚴(yán)重程度采取問責(zé)、處罰直至撤銷其審評資格。其次,建議監(jiān)管部門建立投訴舉報機(jī)制,將社會對第三方機(jī)構(gòu)審評結(jié)果和運(yùn)營機(jī)制的反饋意見作為第三方考評的部分依據(jù)。最后,同行成員和新聞媒體等可加大對第三方審評機(jī)構(gòu)的關(guān)注,及時曝光不良現(xiàn)象,以起到警示作用。這些措施不僅可以增加公眾對醫(yī)療器械行業(yè)的了解,同時可以督促第三方機(jī)構(gòu)嚴(yán)格遵守工作紀(jì)律,保證審評的公正性。