您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2022-10-30 00:12
摘 要 Abstract
目的:通過對國內外已上市連續制造口服固體制劑藥學審評內容的研究,為開展我國連續制造口服固體制劑的藥學審評工作、制定連續制造相關的指導原則或標準、推進ICH《Q13 :原料藥和制劑的連續制造》指導原則在我國的實施與轉化提供科學理論借鑒。方法:在國家藥品監督管理局、美國食品藥品監督管理局、歐洲藥品管理局、日本藥品及醫療器械綜合機構和英國藥品和健康產品管理局的網站,查詢以連續制造生產模式獲批的口服固體制劑審評報告,從中提取與連續制造生產模式相關的藥學審評內容,通過對比分析等研究方法,闡述各國藥品監管機構對連續制造口服固體制劑的藥學審評關注點,并形成我國藥品審評機構未來對此類藥品的藥學審評啟示。結果與結論:連續制造口服固體制劑的藥學審評工作應針對原料藥和輔料、制劑研發、生產工藝和工藝控制、批次描述、工藝驗證、產品放行、穩定性考察、現場檢查、上市后變更等方面進行重點審評。此外,在制定連續制造生產模式相關的指導原則和標準時也應將其考慮在內,以監管促發展,不斷推進我國口服固體制劑的連續制造研發新局面。
Objective: Through the research on the pharmaceutical evaluation content of marketed continuous manufacturing oral solid preparations in China and abroad, this study aims to provide a scientific and theoretical reference for carrying out the pharmaceutical evaluation of continuous manufacturing oral solid preparations in China, formulating guidelines or standards related to continuous manufacturing, and promoting the implementation and transformation of ICH “Q13: continuous manufacturing for drug substances and drug products” guideline in China. Methods: On the websites of National Medical Products Administration, U.S. Food and Drug Administration, European Medicines Agency, Pharmaceuticals and Medical Devices Agency, Medicines and Healthcare products Regulatory Agency, the evaluation reports of marketed continuous manufacturing oral solid preparations were searched, and the pharmaceutical evaluation content related to the continuous manufacturing production mode were extracted. By comparative analysis and other research methods, the concerns of each national medicines regulatory agency on the pharmaceutical evaluation of continuous manufacturing oral solid preparations were elaborated, providing useful experience of pharmaceutical evaluation of such medicines for China’s regulatory agency in the future. Results and Conclusion: The pharmaceutical evaluation of continuous manufacturing oral solid preparations should focus on the aspects of active pharmaceutical ingredients and excipients, preparation research and development, production process and process control, batch description, process validation, product release, stability investigation, on-site inspection, post-approval changes, etc. In addition, it should also be taken into account when formulating the guidelines and standards related to continuous manufacturing production mode, so as to lead its development through supervision, and continuously promote the new situation of research and development of continuous manufacturing oral solid preparations in China.
關鍵詞Key words
連續制造;口服固體制劑;藥學審評;研究與啟示
continuous manufacturing; oral solid preparations; pharmaceutical evaluation;research and insight
基金項目
國家重點研發計劃(2021YFB3201202);山東省重點研發計劃(重大科技創新工程)項目(2021CXGC010507);2021 年中國藥品監督管理研究會課題:緩控釋藥品監管科學研究
1 前言
在全球制藥技術革新和先進制造迅速發展的時代背景下, 連續制造(continuous manufacturing,CM) 作為一種先進的生產模式,可為社會、患者和醫藥企業帶來顯著的效益。原料藥或輔料在工藝起點被連續地送入工藝序列中,在生產過程中發生持續轉化,同時產品在終點被持續輸出。連續制造生產模式具備以下幾大優勢:①生產步驟連續無間歇,消除了傳統批量生產模式中步驟間的停頓,提高了生產效率。②實現了產品質量的實時過程監控,減少了人為判斷錯誤的機會,改善了藥品質量。③生產設備占地面積小,可使用現有的連續制造生產設備快速研發新的工藝過程,降低生產和維護成本,具備生產靈活性和敏捷性。④ 端到端的連續制造生產模式可以顯著加速供應鏈的運行, 并降低存儲和中間運輸成本。⑤實現個性化制造,如3D打印技術。⑥減少了對環境的影響,體現了以綠色制造推動工業高質量發展的理念[1]。
筆者通過查詢國家藥品監督管理局(National Medical Products Administration ,NMPA)、歐洲藥品管理局( European Medicines Agency,EMA)、美國食品藥品監督管理局(Food and Drug Administration,FDA)、日本藥品及醫療器械綜合機構(Pharmaceuticals and Medical Devices Agency,PMDA)、英國藥品和健康產品管理局(Medicines and Healthcare products Regulatory Agency,MHRA)的網站,截至2022 年7 月,共有10 種采用連續制造生產模式的口服固體制劑在這些機構獲得上市許可,其中7 種口服固體制劑得到了多個藥品監管機構的上市許可[2-24]。具體信息見表1。FDA 批準的口服固體制劑數目最多(8 個),其次是EMA(7個)、PMDA(4 個)、NMPA(2個) 和MHRA(1 個)。FDA 對連續制造口服固體制劑的藥學審評工作主要由藥物物質、藥物產品、過程、新興技術、設備、過程分析技術(process analyticaltechnology,PAT)、生物藥劑學、應用技術、環境等審評小組共同完成,為了大力推進連續制造生產模式,FDA 還創建了新興技術計劃,連續制造藥品申請人可通過此計劃,加快此類藥品的獲批上市。EMA 成立了過程分析技術團隊和創新工作組,協助并參與人用藥品委員會的審評工作。PMDA 成立了先進制造技術工作組,協助藥品審評委員會進行審評工作。對于第一個以連續制造生產模式獲批上市的口服固體制劑Orkambi®, 還被EMA 和FDA 列為質量源于設計(quality by design,QbD) 合作試點,通過審評意見征求、審評過程中質量信息和決策信息共享,共同完成了對該制劑的審評工作[25]。


在我國,國際人用藥品注冊技術協調會(ICH)工作辦公室也針對連續制造生產模式成立了專家工作組,在充分討論了ICH《Q13:原料藥和制劑的連續制造》(Q13: Continuous Manufacturing for Drug Substances and Drug Products) 指導原則在我國實施的可行性基礎上, 已完成了眾多的實施轉化工作。2021 年10 月,國家藥品監督管理局藥品審評中心(Center for Drug Evaluation,CDE)發布了《關于公開征求ICH 指導原則〈Q13:原料藥和制劑的連續制造〉意見的通知》[26]。目前, 已有2 種口服固體制劑(Verzenio® 和Cibinqo®) 分別在2021 年和2022 年以連續制造生產模式在我國得到獲批,但是暫無申請上市技術審評報告可循。國內已有眾多醫藥企業著手連續制造生產模式的研發工作,相關審評工作還處于摸索階段。因此,充分借鑒和學習國外連續制造藥品的審評思路,提前明確我國連續制造藥品的審評審批工作重點,制定相關法規、技術指南或標準,優化審評審批制度,對提高我國藥品研發創新能力具有重要意義。
在查詢各國連續制造口服固體制劑的審評報告過程中,筆者發現國外藥品審評機構對連續制造生產模式存在保密機制, FDA和PMDA 審評報告中有眾多覆蓋信息的情況,無法獲得完整的藥學審評內容,這也說明了連續制造是歐美日等發達國家或地區高度重視的先進生產模式,我國更應通過監管努力,鼓勵相關從業人員破解技術封鎖,攻克關鍵技術難題,做好連續制造藥品的研發,使中國的制藥工業不受國外技術限制。
2 國內外已上市的連續制造口服固體制劑的概況
國內外已上市的連續制造口服固體制劑的生產商和生產工藝等情況見圖1。10 種口服固體制劑的劑型均為薄膜包衣片劑,Orkambi®還有顆粒劑型被FDA和EMA 獲批上市。Janssen 公司表示Prezista®連續制造生產線可減少制造和檢驗周期,生產空間明顯減小,在降低生產過程風險的同時還能保證現有產品的質量[27]。表1 涉及的所有藥品批件中有36.4% 以罕見病藥物獲批,研發成本遠高于其他普通口服固體制劑,也采用連續制造生產模式,更加說明連續制造生產模式存在巨大優勢,即采用連續制造生產模式的累積效應可以減少或緩解藥品短缺,增加患者的藥品可及性,提高醫藥企業生產效率和經濟效益。

此外,FDA 近期也回顧了連續制造藥品的申報情況,相比傳統批量生產模式的藥品,連續制造生產模式能獲得更快的批準(圖2)和更高的收入(圖3)。從產品申請到獲批平均快9 個月(中位數為3 個月),從產品提交監管機構到進入市場平均快12 個月( 中位數為4 個月),從產品獲批到進入市場平均快3 個月(中位數為1 個月)。2020 年,連續制造藥品的平均月收入為4290 萬美元,銷售連續制造藥品的公司預計實現了1.71 億~5.37 億美元的早期收入收益。與傳統批量生產模式的藥品申請相比,回顧結果并未表明連續制造藥品申請的監管提交或結果的風險更高,而對于與生產工藝變更或批準前檢查相關的申請內容,也沒有發現實質性的監管障礙[28]。Schaber等[29] 對比了連續制造生產模式與傳統批量生產模式的經濟性問題,發現連續制造生產模式的資本支出會降低20%~76%。


具備如此眾多優勢的連續制造生產模式,是推動我國醫藥產業技術變革和優化升級的重要著力點,我國藥品監管機構可從以下4 個方面重點推進其研發和應用,加快我國由制藥大國向制藥強國的轉變:①鼓勵新藥研發企業基于口服固體制劑品種特點,對于速釋型薄膜包衣片劑或顆粒劑,可優先考慮采用連續制造生產模式,且同一藥品不同規格可采用相近的連續制造研發策略,進而降低生產成本,提高臨床新藥的可及性。②鼓勵國內仿制藥企業,針對臨床應用較廣、產量需求較大的仿制藥品種,進行連續制造生產模式研發,通過上市后變更提高生產效率,滿足臨床的用藥需求。③對于連續制造藥品的審評工作,除了制定相關的法規、技術指南和標準,還應鼓勵醫藥企業就連續制造生產模式研發的合規性,盡早地多次與監管機構溝通交流,使其滿足我國《藥品生產質量管理規范》(Good Manufacturing Practice,GMP)的要求。④在對ICH Q13 指導原則轉化實施的過程中,鼓勵國外以連續制造獲批的藥品在我國注冊上市,以“引進”促“自研”,形成內外循環合力,這些“引進”的連續制造藥品也可通過ICH 與其他國家或地區的監管機構建立共同審評機制。
3 國內外連續制造口服固體制劑的藥學審評內容分析與啟示
10 種連續制造口服固體制劑中9 種是以新藥申報上市,剩下的Prezista®是以補充申請由傳統批量生產模式變更為連續制造生產模式。對于我國連續制造口服固體制劑上市申請來說,主要可分為2 種情況:① 已上市口服固體制劑由傳統批量生產模式變更為連續制造生產模式。②未上市口服固體制劑采用連續制造生產模式申報上市。因此,筆者通過對國內外連續制造口服固體制劑藥學審評內容的研究, 針對上述2 種情況形成藥學審評啟示。
3.1 原料藥和輔料
10 種連續制造口服固體制劑所用原料藥和輔料見表2。通過分析可發現67% 的原料藥具有非吸濕性的特點, 而微吸濕性和吸濕性原料藥僅占其中的25% 和8%。推測連續制造口服固體制劑多采用非吸濕性原料藥的原因, 應是微吸濕性和吸濕性原料藥在儲存和生產過程中會吸收水分子導致自身水解,且加工前還需進行復雜的干燥步驟,非吸濕性原料藥則能避免這些缺點, 具有較強的穩定性。在Symdeko®、Symkevi®、Orkambi®、Trikafta®、Kaftrio®原料藥的生產過程中,還基于研發研究建立了生產過程控制策略,同時,由于這些藥品的原料藥在水中溶解度較差,生產商通過研發噴霧干燥分散體(spray dried dispersion,SDD), 即將溶解度較差的原料藥完全溶解在噴霧干燥溶劑系統中,在與其他原料藥或輔料化學與物理兼容性良好的前提下,以其無定形的中間體用于口服固體制劑的生產,以達到減小顆粒尺寸提高制劑生物利用度的目的。此外, 因替扎卡托和依伐卡托被Vertex 公司在多個規格多個口服固體制劑中使用, 因此在后續制劑的審評中,FDA 提到無需對其進行詳細審查,可依靠已建立和批準的化學成分生產和控制(chemistry manufacturing and controls,CMC)信息來支持后續制劑的原料藥藥學審評。

表2 中的輔料主要包括片芯和薄膜包衣輔料,其中大部分為各國藥典中常用輔料,僅有不到5% 為新型輔料。對各個輔料在10 種連續制造口服固體制劑的使用次數進行排序(圖4),次數靠前的分別為:二氧化鈦、硬脂酸鎂、微晶纖維素、聚乙二醇、羥丙甲纖維素、滑石粉、乳糖、氧化鐵黃、氧化鐵紅和羧甲淀粉鈉等。
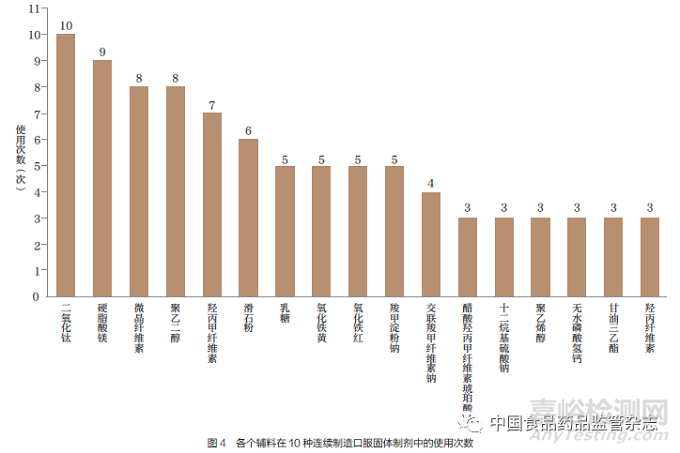
原料藥和輔料是決定藥品質量的關鍵因素,也是連續制造發展的先決條件。目前,我國口服固體制劑的藥學審評工作多關注原料藥和輔料的來源、相關證明文件、執行標準及檢驗報告。對于我國2 種連續制造口服固體制劑上市申請,筆者認為原料藥除了關注非吸濕性特點、水中溶解度較差的原料藥可研發噴霧干燥分散體、推薦《中國藥典》常用輔料,還應基于原料藥和輔料的性質,對其進行除《中國藥典》要求以外的物理屬性評價,如原料藥和輔料的分散性,因為口服固體制劑顆粒粒徑分布、形狀、密度、內聚力等可能會影響連續制造生產時的流動性并形成物料分層。而對于原料藥和輔料本身,可鼓勵使用連續制造生產模式中必備的快速分析工具(過程分析技術),對其進行輸入物料屬性控制和過程控制,從生產源頭和生產過程保證最終制劑的安全性和有效性。此外,在10 種連續制造口服固體制劑的研發過程中,國外制藥企業已基于連續制造生產模式對原料藥和輔料展開研究,間接地也為我國連續制造口服固體制劑的原料藥和輔料的使用提供了參考。CDE 可基于相關課題研究,在評估物料屬性對物料流動、工藝動態和最終產品質量的潛在影響基礎上,制定《連續制造口服固體制劑研發優先使用的原料藥和輔料推薦名單》,表2 中的原料藥和圖4 排序前10 位的輔料可被納入名單考慮范圍內。
3.2 制劑研發
10 種連續制造口服固體制劑的研發都是按照ICH《Q8(R2):藥品研發》[Q8(R2):Pharmaceutical Development] 指導原則的QbD 理念進行的,即質量不是通過檢驗注入產品中,而是通過設計賦予的。與傳統批量生產模式口服固體制劑不同的是,連續制造口服固體制劑的研發必須增加對產品、工藝和過程的理解,并對生產進行全過程的控制和持續改進,具體內容如下。
3.2.1 定義目標產品質量概況(quality target product profile,QTPP)
確定目標產品的用途和預定義產品質量,連續制造口服固體制劑QTPP 普遍包括性狀、鑒別、含量均勻性、有關物質、溶出度等。
3.2.2 概括連續制造生產工藝
主要涉及進料、混合、制粒、干燥、研磨、壓片、包衣等。
3.2.3 理解連續制造工藝知識
基于原料藥和輔料屬性、操作單元、設備能力、規模放大等信息, 通過初始風險評估確定連續制造產品的關鍵質量屬性(critical quality attributes,CQAs)、關鍵工藝參數(critical process parameters,CPPs)或理想生產范圍等。
3.2.4 開展連續制造工藝研發
可借助多變量實驗設計(design of experiments,DOE)、風險分析、生產和控制經驗知識等, 對關鍵工藝參數的正常操作范圍進行深入研究,開展連續制造工藝研發。此外,需要關注到, 在EMA 獲批的Daurismo®借助風險評估指導了停留時間分布(residence time distribution,RTD)的研究;在EMA 獲批的Kaftrio®進行了連續制造生產系統的設計空間范圍研究,可以在批準后進行某些工藝更改(如運行時間、過程參數)以調整輸出,而無需在批準后進行補充申請的材料遞交。
3.2.5 確定連續制造生產過程
基于QbD 或商業規模批次的研究數據,總結原料藥和輔料屬性、連續制造生產工藝的研究結果、保證制劑質量的多維空間等內容,并確定影響制劑關鍵質量屬性的因素,確保所有關鍵質量屬性都在可接受范圍內。
3.2.6 確定連續制造生產控制策略
保證中間控制(in processcontrol,IPC)參數、關鍵關鍵工藝參數、關鍵質量屬性等都在一個可接受的范圍內上下波動。此外,對于生產期間發生的瞬態事件,如計劃內的工藝啟動、關閉和暫停或計劃外的擾動,也都應有相應的過程分析技術對其進行實時檢測,且過程分析技術的測量頻率足以檢測擾動,能通知工藝調整并確保根據預定標準及時分流物料。例如,在EMA 獲批的Symkevi® 和Orkambi®通過過程分析技術監測連續制造口服固體制劑的生產過程,減輕了擾動對最終制劑質量的影響。
3.2.7 持續改進連續制造產品質量
基于對產品和工藝知識的理解、質量風險管理(如原輔料屬性、因清潔不當造成的交叉污染)等,對生命周期管理提出意見并改進,保證產品質量持續穩定和均一。例如,在EMA 獲批的Verzenio®和Kaftrio®則運用ICH《Q12 :藥品生命周期管理的技術和監管考慮》(Q12 :Techinical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management) 指導原則中的批準后變更管理方案(post-approval change management protocol,PACMP),完成了生產場所等的變更和技術轉移,同時基于臨床數據獲得更高水平的工藝理解和驗證。
對于我國連續制造口服固體制劑研發的藥學審評,應重點關注對制劑的產品理解、工藝與過程理解和過程控制策略制定,因此與其相關的工藝研發、產品設計、裝備改造、過程控制內容應在申報資料中被充分闡述。對于未上市口服固體制劑采用連續制造生產模式申報上市的情況,可基于上文總結的7 部分進行相應的審查,確保申請人已經完成了充分的研究。對于已上市口服固體制劑由傳統批量生產模式變更為連續制造生產模式的情況,因過去已有相關的研發經驗,因此可以在對過往注冊申報材料審查的基礎上,重點關注生產工藝的改進和優化工作,如描述工藝研究的主要變更(批量定義、連續制造設備、過程分析技術監測點和分析模型、關鍵工藝參數等),建立覆蓋整個制劑生命周期的質量管理體系。
3.3 生產工藝與工藝控制
在EMA 獲批制劑的審評報告中,出現了“全部生產環節連續的生產模式”和“部分生產環節連續的生產模式”的概念,具體生產工藝見圖5。采用全部生產環節連續的生產模式的制劑有Orkambi®、Tramacet®、Symkevi ®、Prezista®,其中Orkambi®既有全部生產環節連續的生產模式,又有部分生產環節連續的生產模式。對于部分生產環節連續的生產模式,連續制造生產環節情況如下:①從預混到壓片環節。②從進料到壓片環節。③從進料到包衣環節。④從制粒到研磨環節。⑤從制粒到干燥環節。⑥從制粒到壓片環節。⑦從進料到包裝環節。此外, 在EMA 獲批的Daurismo®和Cibinqo®均采用了輝瑞公司研發的便攜式、連續式、微型和模塊化連續制造系統,該系統主要執行成品制造、質量控制測試、初級和次級包裝以及批次放行等功能。

10 種連續制造口服固體制劑工藝控制策略主要包括對原輔料的屬性判斷與控制、單元操作控制和集成操作控制、工藝設計空間參數監控、以過程分析技術為主要方法的過程控制。這些控制策略涉及每種物料、工藝步驟和連續制造設備,能夠實時處理擾動或發出異常警報。其中以過程分析技術為主要方法的過程控制是連續制造生產模式區別于傳統批量生產模式的地方,過程分析技術是指對制劑生產過程中關鍵質量屬性或影響關鍵質量屬性的物料參數進行檢測,該技術既可以分析出因生產工藝引起的質量波動,隨時保證工藝性能和產品質量,又可以利用其變異性分析生產工藝在設計空間運行的能力,為檢測抽樣計劃和頻率提供依據,因此被大多連續制造口服固體制劑采用。
以在EMA 獲批的Symkevi®為例,繪制了該制劑的連續制造平臺簡圖(圖6),主要包括原輔料、工藝設備、工藝流程、5 個過程分析技術監測點、工藝參數、2個不合格顆粒/ 片芯分離點、3 個停留時間分布研究點、全流程實時放行檢測(real time release testing,RTRT)等。連續制造平臺控制系統可顯示正在運行的單元操作、過程控制和關鍵工藝參數的信息,實時監控工藝參數和設計空間范圍,并在出現偏差時發出警報。針對整個連續制造控制平臺,還考察了啟動、關閉、過程暫停前后對產品質量的影響,并獲得了目標產品質量的波動范圍。其停留時間分布研究是通過產品關鍵要素(product key,PK)對噴霧干燥分散體、輔料、包衣懸浮液進行跟蹤研究,產品關鍵要素是可以被分離剔除的顆粒或片芯的最小不合格單位,隨后基于停留時間分布研究結果和工藝設備進行綜合考慮來設計分離點和分離方法。該制劑也基于全流程RTRT 制定了應急策略,用以規定RTRT 設備發生故障時臨時使用的替代測試或監測方法。

此外,對于工藝控制相關的模型審評,主要重點關注以下3類:①過程分析技術模型。②停留時間分布研究模型。③綜合控制模型,如傳感器控制模型等。以過程分析技術的模型審評為例,表3 整理了3 種類型的過程分析技術,其中光譜技術模型在連續制造生產模式中的研究和應用最為廣泛。基于圖5 的梳理,10 種連續制造口服固體制劑存在部分相同的工藝環節,故圖7 對關鍵生產環節的生產設備和過程分析技術監測點進行總結,可見原料藥含量是過程分析技術監測的重點,而近紅外光譜技術多次被應用于原料藥含量、混合均勻度等的分析。FDA 和EMA 要求近紅外光譜模型的研發應當遵循基于科學和風險的原則,包括風險評估和實驗設計研究,以確定哪些變量可能會影響近紅外光譜模型的建立。注冊申報時需提供近紅外光譜儀、軟件、光纖探頭等信息;近紅外光譜化學計量學模型的研發需根據FDA 的《化學藥品與生物制品的分析程序與方法驗證》(Analytical Procedures and Methods Validation for Drugs and Biologics)或EMA 的《制藥工業近紅外光譜技術應用、申報和變更資料要求指南》(Guideline on the Use of Near Infrared Spectroscopy by the Pharmaceutical Industry and the Data Requirements for New Submissions and Variations),充分描述分析流程和驗收標準等,確保該模型的專屬性和穩健性。此外,還需參考ICH Q12 指導原則提出生命周期管理的模型維護程序。


對于我國的2 種連續制造口服固體制劑上市申請,圖5 的連續濕法制粒、連續干法制粒、連續直接壓片都是可以接受的連續制造生產模式,其中,連續直接壓片更值得推薦,這與其工藝流程短、易控制的特點密不可分。圖5 中出現的全部生產環節連續的生產模式和部分生產環節連續的生產模式,更是說明不包含制劑全部生產環節的連續制造生產線也是可以被審評機構所接受的。因此,我國藥品監管機構在連續制造相關指導原則或標準的制定上,可鼓勵醫藥企業基于現有的生產設備和生產條件,確定是否研發全部生產環節連續的生產模式,無條件研發的企業,可從部分生產環節連續的生產模式入手,先實現涵蓋2~5 個生產環節的連續制造生產線的研發或改造,階段性地向全部生產環節連續的生產模式靠攏。工藝控制方面,除了與連續制造相關的工藝流程圖、工藝描述、主要生產設備、商業生產規模與依據,審評方面還應當重點審評連續制造生產模式所特有的控制策略,如以過程分析技術為主要方法的過程控制,審查申請人是否針對制劑生產形成了“物質基礎正確、過程控制準確、體系設計科學”的連續制造控制策略,圖6 的連續制造平臺和圖7 總結的過程分析技術監測點都是藥學審評過程中可借鑒的工藝控制案例。而對于過程分析技術本身,如近紅外光譜技術,應重點評估是否對分析程序和方法驗證等進行了充分的研究和描述,并形成相關的驗收標準。
3.4 批次描述
在10 種連續制造口服固體制劑的藥學審評報告中,均定義了預期的商業規模批量大小。但對批次的具體描述內容較少,只有在EMA 獲批的Daurismo®和Cibinqo®、在PMDA 獲批的Tazverik® 提到批量大小取決于生產預定數量片芯所需的原料藥質量,在FDA 獲批的Verzenio®定義“批量”概念時考慮了所采用的原料藥的質量和連續制造質量流量,如其他參數不變,可通過延長生產時間增加批量。在EMA 獲批的Kaftrio® 還提到批量大小的具體數值(60kg)。
生產批次的正確劃分是確保制劑受控、可追溯的必要條件,也是申請人能夠開展質量管理和生產管理的前提要求。我國《藥品生產質量管理規范》(2010 年版) 在借鑒EMA 的GMP 時,也在術語中保留了EMA 對連續制造藥品的一些特殊考慮,如第三百一十二條:“在連續生產情況下,批必須與生產中具有預期均一特性的確定數量的產品相對應,批量可以是固定數量或固定時間段內生產的產品量。”因此,對于我國的2 種連續制造口服固體制劑上市申請,均應考慮連續制造系統的開機、受控狀態、調整、停機等生產狀態的生產能力和系統產生的廢料,給出定義批量的方法,并能充分證明連續制造生產系統生產一致批量大小產品的穩健性。由于制劑批次還具體涉及QbD 研發批次、臨床批次、注冊批次和商業規模生產批次,因此需要確定好各自的批量數值或范圍,合理并清楚地描述出每種批次的情況。
3.5 工藝驗證
工藝驗證內容多出現在EMA公布的審評報告中,此外EMA曾發布過《成品工藝驗證指南——監管提交文件的信息和數據要求》(Guideline on Process Validationfor Finished Products—Information and Data to be Provided in Regulatory Submissions),基于這份指南,Symkevi®、Kaftrio® 采用了持續工藝確認(continuous process verification,CPV)方案對工藝進行驗證。而其他制劑則采用傳統工藝驗證方法,除了其中的Kaftrio®和Verzenio®分別采用2 個和12 個商業規模批次進行工藝驗證,剩余的則采用了連續3 個商業規模批次進行工藝驗證。對比連續制造生產模式和傳統批量生產模式,工藝驗證的區別在于需要根據驗證總體規劃對過程控制性能進行評估。
對于我國的2 種連續制造口服固體制劑上市申請,鼓勵申請人可采用持續工藝確認的方法在擬定的商業規模生產場地完成工藝驗證,將工藝研發、商業生產工藝驗證、商業規模生產中的持續工藝確認相結合,確定工藝始終如一的處于受控狀態,實現制劑的生命周期管理,隨著生產商對工藝的理解和工藝性能控制水平不斷提高,還可對持續工藝確認的范圍和頻率進行周期性的審核和調整。工藝驗證資料至少應當包括工藝驗證方案、工藝驗證報告、批生產記錄樣稿等。工藝驗證內容應當包括批號、批量、連續制造設備的選擇與評估、連續制造工藝條件、連續制造工藝參數和工藝參數可接受范圍、過程控制分析方法、抽樣方法及計劃、連續制造工藝步驟評估、不合格物料的分離能力等。
3.6 產品放行
10 種連續制造口服固體制劑大部分是通過傳統的成品測試進行產品放行, 其中也不乏采用RTRT 進行產品放行, 如在EMA 獲批的Orkambi®、Symkevi®、在EMA 和FDA 獲批的Verzenio®。對產品放行檢測項目進行梳理(表4),申請人主要進行了外觀、含量測定、鑒定、溶出度、劑量單位均勻度、微生物限度及降解產物等項目的檢測。在PMDA 獲批的Tazverik®還采用相同的取樣點, 對連續制造和傳統批量生產模式的產品放行檢測結果進行對比,分析結果顯示無明顯差異。此外,在EMA 和FDA 獲批的Symkevi®/Symdeko®、在EMA 獲批的Orkambi®還建立了RTRT 溶出度模型,并研究證明了RTRT溶出度模型能夠表征原料藥溶出度的能力,具體內容見表5。RTRT 技術被醫藥企業視為連續制造商業化后獲得高效率與經濟效益的重要手段,同時EMA 和FDA 還鼓勵實施部分或全部產品質量屬性的RTRT,這使該技術在產品放行中得以應用和推廣。例如,EMA 發布了《實時放行檢測指南(先前為參數放行指南)》[Guideline on Real Time Release Testing (Formerly Guideline on Parametric Release)], 鼓勵使用近紅外光譜技術和拉曼光譜技術作為RTRT 工具,通過與《歐洲藥典》(European Pharmacopoeia)中常規分析方法[ 如高效液相色譜法(high performance liquid chromatography,HPLC)] 的結果比較,完成RTRT 方法的研發和驗證。EMA 也提出在口服固體制劑商業生產的第一年,每次生產活動至少有1 個批次,之后每年至少有1 個批次,進行產品放行的平行檢測,以研究近紅外光譜模型或拉曼光譜模型用于RTRT 的穩定性。當RTRT 不能使用或需要系統更新時,則以常規分析方法代替。


目前,我國暫無與產品放行直接相關的指導原則,醫藥企業多參考ICH《Q6A :質量標準:新原料藥和新藥制劑的檢測方法和可接受標準:化學物質》(Q6A:Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances)、《Q3B(R2):新藥制劑中的雜質》[Q3B(R2): Impurities in New Drug Products] 等指導原則中的內容確定適當的抽樣計劃和放行標準。對于我國的2 種連續制造口服固體制劑上市申請,應提供至少3 個商業規模批次的產品放行報告。基于表4 的梳理,在制定產品放行的質量標準時應重點考慮外觀、含量測定、鑒定、溶出度、劑量單位均勻度、微生物限度及降解產物。此外,還應考慮對水分和粒度分布的檢測,若具有單獨的放行標準和貨架期標準也應分別說明。對于已上市口服固體制劑由傳統批量生產模式變更為連續制造生產模式,應進行連續制造和傳統批量生產模式的產品放行對比,以證實連續制造生產模式可以制造出與傳統批量生產模式質量一致或更高的藥品。此外,對于口服固體制劑產品放行的重要檢測項目溶出度來說,我國CDE 已發布《普通口服固體制劑溶出度試驗技術指導原則》。該指導原則還針對口服固體制劑的處方工藝在批準后發生變更時,如何通過溶出度試驗確認口服固體制劑質量和療效的一致性提出了建議,可用于指導連續制造口服固體制劑的藥學研究工作。對于我國的2 種連續制造口服固體制劑上市申請,若按《中國藥典》的溶出度方法進行溶出度表征,應建立溶出度標準的方法和溶出曲線比較的統計學方法,根據可接受的臨床試驗用樣品、生物利用度和(或)生物等效性試驗用樣品的溶出度結果,制定溶出度標準并提供相關檢測數據。對于已上市口服固體制劑由傳統批量生產模式變更為連續制造生產模式,還應提供連續制造生產批次和過去的傳統批量生產批次的溶出度對比數據,確認產品質量和性能是否保持一致。而多規格采用連續制造生產模式的口服固體制劑,溶出度比較試驗可用于申請小劑量規格制劑體內生物等效性試驗的豁免。在此基礎上,若溶出度的表征是采用類似表5 的RTRT 溶出度模型,或是在產品放行中引入RTRT 技術,都應當提供完整的模型研發和校準的原始建模數據,包括但不限于程序文件、模型研發、校準和方法學驗證的輸入和輸出數據;提供用于模型研發、校準和驗證的各個批次或環節的配方和工藝參數信息;做好產品生命周期管理的模型更新和完善工作,確保該檢測技術的穩健性;并以臨床相關性數據驗證該技術的應用能力。
3.7 穩定性考察
對于10 種連續制造口服固體制劑的穩定性考察,主要包括影響因素試驗(表6)、加速試驗和長期試驗(表7)以及其他試驗(表6)。影響因素試驗中主要對光、濕、熱、酸、堿、氧化等因素進行考察,部分制劑根據加速試驗結果,增加了中間條件試驗(表8)。據此為其處方、工藝、包裝、貯藏條件、復驗期和有效期的確定提供了支持性信息。同時,簡略設計方法(括號法和矩陣法)也被用于穩定性考察。值得關注的是,在FDA 和EMA 獲批的Symdeko®/Symkevi®和在PMDA 獲批的Tazverik®還對傳統批量生產批次和連續制造生產批次的穩定性結果進行了對比,證明了連續制造的生產能力。
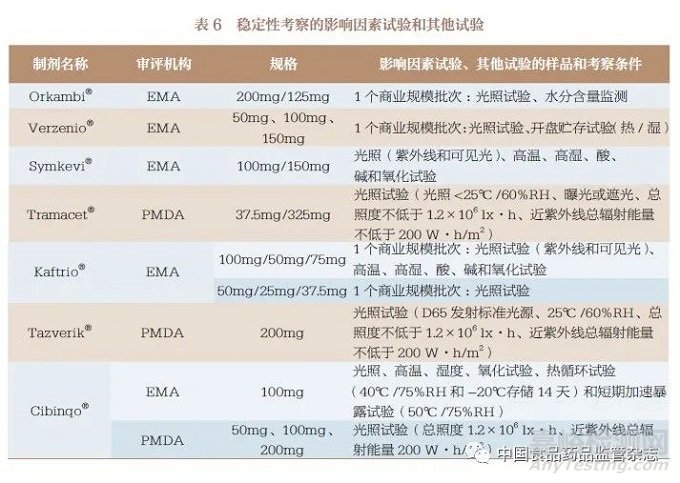


針對穩定性考察,我國CDE已經發布了《化學藥物(原料藥和制劑)穩定性研究技術指導原則》等,同時也有ICH《Q1 :穩定性》(Q1: Stability)指導原則可參考。對于已上市口服固體制劑由傳統批量生產模式變更為連續制造生產模式的情況,應提交3個傳統批量生產批次和3 個連續制造生產批次的穩定性考察對比結果。對未上市口服固體制劑采用連續制造生產模式申報上市的情況,應提交至少3 批中試規模批次或驗證批次的穩定性考察結果;對于中試規模批次或驗證批次,其合成路線、處方及生產工藝應與商業化規模產品一致或與商業化規模產品的關鍵工藝步驟一致,且各批次樣品質量應能代表商業化規模產品的質量,包裝容器也應與商業化規模產品相同或相似。基于表6~ 表8 的梳理,影響因素試驗和其他試驗可采用1 個注冊批次的樣品,加速試驗(40℃ ±2℃ /75%RH±5%RH)和必要的中間條件試驗(30℃ ±2℃ /65%RH±10%RH)應采用3 個注冊批次的樣品進行至少6 個月的考察, 長期試驗(25℃±2℃/60%RH±10%RH或30℃ ±2℃ /65%RH±10%RH)應采用3 個注冊批次的樣品進行至少12 個月的考察。如果以上幾種試驗結果有不明確的情況,應加試同樣批次量的樣品,其他考察條件、分析方法和考察指標根據藥物性質進行確定。此外,產品被CDE 獲批上市后,應承諾以上市后生產的前3 批產品進行長期留樣穩定性考察,并對每年生產的至少1 批產品進行長期留樣穩定性考察。
3.8 現場檢查
現場檢查內容多出現在FDA公布的審評報告中, 在FDA 獲批的Orkambi®、Verzenio®、Daurismo®均進行了批準前現場檢查,483 缺陷報告中列出了申請人質量體系不符合GMP 的情況,具體內容見表9,主要涉及生產工藝和控制方面等內容。

對于我國的2 種連續制造口服固體制劑上市申請,實施現場檢查工作時,除了對處方與工藝研究、樣品試制、技術轉移、質量管理、穩定性研究、數據可靠性、生產現場的真實性等常規檢查,更應注重連續制造商業規模生產過程和質量控制策略是否與申報資料保持一致,如廠房設施、連續制造關鍵設施設備和物料、連續制造生產系統、連續制造標準操作規程、批生產完整記錄、連續制造工藝驗證方案及報告、質量標準、過程分析技術與分析模型、不合格物料的分離能力等,同時檢查連續制造商業規模生產條件是否具備連續制造的生產能力,保證持續輸出質量穩定和均一的制劑產品。對于已上市口服固體制劑由傳統批量生產模式變更為連續制造生產模式,由于該變更屬于重大變更的情形,現場檢查還應當調取先前的現場檢查結果,考察既往是否存在重大不合規問題,在此次現場檢查中是否對此作出相應的改進。
3.9 上市后變更
與10 種連續制造口服固體制劑藥學審評相關的上市后變更主要分為3 種:原料藥相關的變更、生產過程相關的變更、制劑產品相關的變更,具體內容見表10。此外,對于連續制造這種特殊生產模式,部分制劑還要求提交與過程控制變更管理相關的批準后變更管理方案。
藥品上市后變更管理屬于藥品生命周期管理的一部分,對于我國的2 種連續制造口服固體制劑上市申請來說,已上市口服固體制劑由傳統批量生產模式變更為連續制造生產模式本身就是一種重大的上市后變更情況,此外便是獲批上市后又出現的原料藥、生產過程和制劑產品相關的變更;未上市口服固體制劑采用連續制造生產模式申報上市獲批后也會存在原料藥、生產過程和制劑產品相關的變更。基于上述變更情況的藥學審評工作,由于我國開展上市后藥學變更審評工作時間相對較短,可借鑒表10中出現的與連續制造藥學審評相關的上市后變更情況,鼓勵申請人在對制劑及其工藝、質量控制等不斷深入理解的基礎上,采用ICH《Q10:藥品質量體系》(Q10:Pharmaceutical Quality System)、《Q12 :藥品生命周期管理的技術和監管考慮》等指導原則的變更管理工具,對變更進行重大、中等和微小的確定并展開相關研究。CDE 可基于申請人提交的補充申請、備案或年度報告材料,不斷積累審評審批經驗,可在借鑒EMA 和FDA 的批準后變更管理方案基礎上,形成與連續制造生產模式相關的上市后變更案例,供相關醫藥企業參考,促進連續制造生產模式的持續改進和創新。

上文主要從原料藥和輔料、制劑研發、生產工藝與工藝控制、批次描述、工藝驗證、產品放行、穩定性考察、現場檢查和上市后變更9 個方面對國外連續制造口服固體制劑藥學審評內容的啟示進行了闡述,此外還應關注到,在NMPA 或其直屬單位已發布的各項指導原則、指南或標準中,暫無直接與連續制造生產模式相關的內容。若要推進連續制造口服固體制劑的研發與上市,完善藥品監督管理政策和技術指導文件是排在首位的工作。因此,建議在現有藥品監管法律法規框架下,明確監管路徑,可基于上文各個藥學審評方面的啟示,陸續制定或完善與連續制造生產模式相關文件(表11)、GMP 附錄或將其收載入《中國藥典》。

4 結論
隨著全球連續制造口服固體制劑不斷獲批上市,連續制造生產模式已成為美歐日等發達國家或地區高度重視的新興技術。在新冠肺炎疫情全球大流行的環境下,藥品緊缺問題突出,連續制造生產模式因其生產速度快、質量穩定、綠色環保等優點使其具有更特殊且重要的意義。藥品生產技術的更新換代速度和醫藥行業快速發展需求,給我國藥品審評機構、藥品審評能力和藥品審評體系現代化建設提出了新的挑戰。本文通過對國內外已上市連續制造口服固體制劑的藥學審評內容進行研究,形成了我國藥品監管機構未來對此類制劑的藥學審評啟示。建議具有前瞻性的連續制造指導原則或標準陸續展開研究、制定、征求意見和發布,起到規范連續制造生產模式和指導其應用的效果,不斷推進ICHQ13 指導原則在我國的實施與轉化。鼓勵有計劃實施連續制造生產模式的醫藥企業,盡早地同CDE 進行溝通交流,提前規避和解決連續制造生產模式合規性和法規適用性的問題。我國藥品監管機構應堅持國際化監管視野,不斷提升我國藥品監管能力,以審評工作促進連續制造藥品的研發,促進安全、有效、穩定、均一和經濟的連續制造藥品上市,建立健全現代化藥品審評體系,增強我國在國際藥品監管領域的話語權,加快我國醫藥產業的高質量發展。
引用本文
孫鐘毓,林泊然,李爽爽,梁夢穎,王浩偉,陳貴鑫 張惠,羅蘇秦*,臧恒昌*.國內外已上市連續制造口服固體制劑藥學審評內容的研究與啟示[J].中國食品藥品監管.2022.09(224):54-77.

來源:中國食品藥品監管雜志


