您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2021-07-04 22:58
分子診斷技術是用分子生物學方法針對人體及各種病原體的遺傳物質的表達及結構進行檢測,從而達到預測及診斷疾病的目的。
近年來,隨著分子診斷技術的升級迭代,分子診斷的臨床應用越來越廣泛和深入,分子診斷市場進入快速發展期。
筆者總結市場上常見的分子診斷技術,分為上中下三篇,上篇介紹PCR技術,中篇介紹核酸等溫擴增技術,下篇介紹測序技術。
01、上篇:PCR技術
PCR技術
PCR (polymerase chain reaction)聚合酶鏈式反應,是體外DNA擴增技術之一,至今已經超過30年的歷史。
PCR技術于1983年由美國Cetus公司的KaryMullis首創,1985 年Mullis申請了PCR專利,同年在Science上發表了第一篇PCR 學術論文,1993 年Mullis因此獲得了諾貝爾化學獎。
PCR基本原理
PCR可以將目標DNA片段擴增一百萬倍以上,其原理是在DNA聚合酶催化下,以母鏈DNA為模板,以特定引物為延伸起點,通過變性、退火、延伸等步驟,體外復制出與母鏈模板DNA互補的子鏈DNA的過程。

標準 PCR 過程分為三步:
1.變性(Denaturation):利用高溫使DNA 雙鏈分離。DNA雙鏈之間的氫鍵在高溫下(93- 98℃)被打斷。
2.退火(Annealing):在 DNA 雙鏈分離后,降低溫度使得引物可以結合于單鏈DNA 上。
3.延伸(Extension):DNA 聚合酶由降溫時結合上的引物處開始沿著DNA 鏈合成互補鏈,延伸完成,則完成一輪循環,DNA片段數增加一倍。
往復循環這三個步驟25-35 次,DNA 片段數將得到指數級增加。

PCR的巧妙之處在于針對不同的目標基因可以設計不同的引物,使目標基因片段在短時間內得到百萬級的放大。
目前為止,PCR可以分為三類,分別是普通PCR、熒光定量PCR和數字PCR。
第一代普通PCR
采用普通PCR 擴增儀來對靶基因進行擴增,然后采用瓊脂糖凝膠電泳對產物進行檢測,只能做定性分析。
第一代PCR主要缺點:
容易發生非特異性擴增和假陽性結果。
檢測耗時長,操作繁瑣。
只能做定性檢測。
第二代熒光定量PCR
熒光定量PCR(Real-Time PCR),也叫做qPCR,通過在反應體系中加入能夠指示反映進程的熒光探針,通過熒光信號的積累來監測擴增產物的積累,通過熒光曲線來判斷結果,并可以借助Cq 值和標準曲線來定量。
qPCR 技術由于操作過程在封閉體系中進行,降低了污染概率,并且可以通過對熒光信號監測從而進行定量檢測,因此臨床應用最為廣泛,已成為PCR中的主導技術。
實時熒光定量PCR所使用的熒光物質可分為:TaqMan熒光探針、分子信標和熒光染料。
1)TaqMan熒光探針:
PCR擴增時在加入一對引物的同時加入一個特異性的熒光探針,該探針為一寡核苷酸,兩端分別標記一個報告熒光基團和一個淬滅熒光基團。
探針完整時,報告基團發射的熒光信號被淬滅基團吸收;PCR擴增時,Taq酶的5'-3'外切酶活性將探針酶切降解,使報告熒光基團和淬滅熒光基團分離,從而熒光監測系統可接收到熒光信號,即每擴增一條DNA鏈,就有一個熒光分子形成,實現了熒光信號的累積與PCR產物形成完全同步。
2)SYBR熒光染料:
在PCR反應體系中,加入過量SYBR熒光染料,SYBR熒光染料非特異性地摻入DNA雙鏈后,發射熒光信號,而不摻入鏈中的SYBR染料分子不會發射任何熒光信號,從而保證熒光信號的增加與PCR產物的增加完全同步。SYBR僅與雙鏈DNA進行結合,因此可以通過溶解曲線,確定PCR反應是否特異。

3)分子信標
是一種在5和3末端自身形成一個8個堿基左右的發夾結構的莖環雙標記寡核苷酸探針,兩端的核酸序列互補配對,導致熒光基團與淬滅基團緊緊靠近,不會產生熒光。

PCR產物生成后,退火過程中,分子信標中間部分與特定DNA序列配對,熒光基因與淬滅基因分離產生熒光。

第二代PCR主要缺點:
靈敏度還有欠缺,低拷貝標本檢測不準確。
存在背景值影響,結果易受干擾。
當反應體系中有PCR抑制物時,檢測結果易受干擾。
第三代數字PCR
數字PCR(DigitalPCR, dPCR, Dig-PCR)通過終點檢測計算目標序列的拷貝數,無需采用內參和標準曲線即可進行精確的絕對定量檢測。
數字PCR采用終點檢測,不依賴于Ct值(循環閾值),所以數字PCR反應受擴增效率的影響降低,對PCR反應抑制物的耐受能力提高,具有很高的準確度和重現性。
由于具備高靈敏度、高精確度的特點,不易被PCR反應抑制劑干擾,無需標準品可實現真正意義的絕對定量,而成為研究和應用熱點。
根據反應單元的不同形式,主要可分為微流體式、芯片式和微滴式三大類系統。
1)微流體數字PCR,Microfluidic digital PCR,mdPCR。
基于微流控技術,對DNA模板進行分液,微流控技術能實現樣品納升級或更小液滴的生成,但液滴需要特殊吸附方式再與PCR反應體系結合,mdPCR已逐漸被其他方式取代。
2)微滴數字PCR,Droplet-based digital PCR,ddPCR。
利用油包水微滴生成技術對樣品進行微滴化處理,將含有核酸分子的反應體系分成成千上萬個納升級的微滴,其中每個微滴或不含待檢核酸靶分子,或者含有一個至數個待檢核酸靶分子。
以伯樂的ddPCR為例,主要包括三個步驟:

第一,準備樣本和生成微滴,如下圖,8x3的微孔板,一排加樣本,一排加油滴,通過微滴生成器每個樣本生成20000個微滴。


第二,進行“油包水”PCR,把微孔板放入PCR擴增儀,對每個微滴進行40個熱循環的PCR反應。
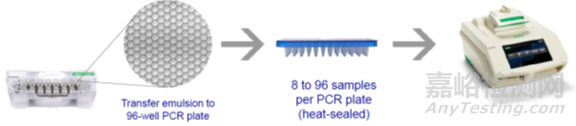
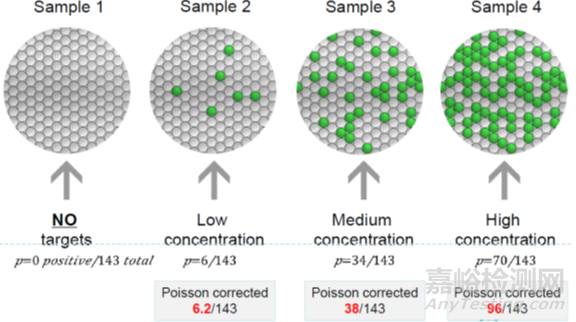
第三,讀取微滴結果,把微孔板放入讀取儀,通過流式細胞技術獲取每個微滴PCR終點結果的熒光信號,并用泊松分布原理糾正結果。
Bio-rad去年上市的Bio-Rad QX ONE數字PCR系統整合了微滴生成、熱循環反應及微滴讀取等步驟,最大程度減少人工操作。
配備四個獨立的熒光檢測通道,結合ddPCR特有的高階多重PCR技術,可以在單反應孔中實現8重拷貝數變異(CNV)檢測、5重突變檢測或4重基因表達檢測。

3)芯片數字PCR,Chip-baseddigital PCR,cdPCR。
利用集成流體通路技術在硅片或石英玻璃上刻上許多微管和微腔體,通過不同的控制閥門控制溶液在其中的流動,將樣本液體分成大小一致的納升級于反應孔種進行數字PCR反應,實現絕對定量。
以法國Stilla technologies 公司的cdPCR技術為例,主要包括以下步驟:
第一,加樣和生成微滴:將樣本和PCR反應液加入微流控芯片,放入儀器中,儀器可通過物理方法生成單層平鋪的20000~30000個微滴陣列。
第二,對每個微滴進行PCR擴增:在Naica Geode微滴生成擴增系統自動進行PCR擴增。
第三,讀取和分析結果:將芯片置于Prism微滴讀取分析系統上進行熒光信號采集,計數陰陽性微滴,通過泊松分布計算獲得靶標基因絕對拷貝數濃度。

總結一下,將樣本和PCR反應液加入微流控芯片,放入儀器中,通過物理方法生成單層平鋪的微滴陣列,隨后對每個微滴進行擴增,然后進行六通道熒光信號采集,計數陰陽性微滴,通過泊松分布計算獲得靶標基因絕對拷貝數濃度。
第三代PCR主要缺點:
儀器和試劑昂貴。
模板質量要求較高,模板量超過微體系量將導致無法定量,過少則定量準確度降低。
當存在非特異性擴增時也會產生假陽性。
PCR的各種延伸技術
1.遞減PCR(touchdown PCR):前幾循環溫度逐漸下降。
2.逆轉錄PCR(RT-PCR):以由mRNA逆轉錄而來的cDNA 為模板,也因為是從表現型基因來進行增量的,由此產生出來的cDNA 產物不帶有內含子(基因中不具意義的段落),常應用于分子克隆技術。
3.熱啟動PCR(hotstart PCR):以高熱激活型核酸聚合酶進行反應,減少非專一性產物。
4.巢式PCR(nested PCR):先用低特異性引物擴增幾個循環以增加模板數量,再用高特異性引物擴增。
5.多重PCR(multiplex PCR):在同一個管中使用多組引物。
6.復原條件PCR(reconditioning PCR):PCR 產物稀釋 10 倍后重新放入原濃度的引物和dNTP 等循環 3 次,以消除產物中的異二聚體。
7.dsRNA 合成(dsRNA replicator):合并使用high-fidelity DNA polymersae、T7RNA聚合酶與Phi6 RNA replicase;從雙股 DNA轉錄為對應的雙股RNA(dsRNA)。可應用于RNAi實驗操作。
8.COLD-PCR (co-amplification at lower denaturation temperature PCR):用以檢測突變或特殊等位基因的PCR 應用技術。
參考文獻:
1.劉婉彤等,分子診斷技術的臨床應用進展
2.楊柳等,非鱗非小細胞肺癌基因檢測技術研究進展
3.G.Terrance Walker,etc.Strand displacement amplification-an isothermal, in vitro DNA amplification technique
3.各公司官網
02、中篇:核酸等溫擴增技術
本篇介紹一下等溫擴增技術。
PCR是使用最為廣泛的核酸擴增技術,以其靈敏性、特異性得到廣泛應用,然而PCR需要反復的熱變性,無法擺脫依賴儀器設備的局限,從而限制了其在臨床現場檢測中的應用。
自20世紀90年代初,很多實驗室開始發展無需熱變性的恒溫擴增技術,現已開發出環介導等溫擴增技術、鏈替代等溫擴增技術、滾環等溫擴增技術、依賴核酸序列等溫擴增技術等技術。
環介導等溫擴增
環介導等溫擴增(loop-mediated isothermal amplification,LAMP)是由日本榮研公司的Notomi等于2000年首先提出來的新的核酸擴增技術。
基于該技術,榮研還曾和國內某公司引起專利糾紛。
其擴增原理是基于DNA在65℃左右處于動態平衡狀態,任何一個引物向雙鏈DNA的互補部位進行堿基配對延伸時,另一條鏈就會解離,變成單鏈。
DNA在此溫度下利用4條特異性引物依靠一種鏈置換DNA聚合酶,使鏈置換DNA的合成不停地自我循環。
先確定目標基因上的6個特異性區域F3、F2、F1、B1、B2、B3,然后依據這6 個特異性區域設計4條引物(如下圖):
正向內引物(forwardinner primer,FIP),由F1c 和F2 組成。
反向內引物(backwardinner primer,BIP),由B1c 和B2 組成,中間均以TTTT作為間隔。
外引物F3、B3 分別由目的基因上的F3、B3 區域組成。

在LAMP 反應體系中,內引物的濃度幾倍于外引物的濃度。內引物先與模板鏈結合合成互補鏈,形成DNA雙鏈。隨后外引物再與該模板鏈結合,形成DNA雙鏈,在BstDNA 聚合酶的作用下,釋放出由內引物合成的互補鏈,該互補鏈經過一系列反應最終形成具有啞鈴結構的DNA單鏈。
以啞鈴結構DNA單鏈自身為模板不斷形成一端開口的過渡性莖環結構DNA,由內外引物引導過渡性莖環結構DNA不斷發生鏈置換延伸反應,最后形成具有多個莖環結構的長度不一的DNA混合物。

環介導等溫擴增的優勢與不足
LAMP 的優勢:
(1)擴增效率高,能夠在1h內有效的擴增1-10個拷貝的目的基因,擴增效率為普通PCR的10 倍-100 倍。
(2)反應時間短,特異性強,不需要特殊的設備。
LAMP 的不足:
(1)對引物的要求特別高。
(2)擴增產物不能用于克隆測序,只能用于判斷。
(3)由于其敏感性強,容易形成氣溶膠,造成假陽性,影響檢測結果。
鏈置換擴增
鏈置換擴增(strand displacement amplification,SDA)是美國學者Walker于1992年首次提出的一種基于酶促反應的DNA體外等溫擴增技術。
SDA 的基本系統包括一種限制性核酸內切酶、一種具有鏈置換活性的DNA聚合酶、兩對引物、dNTP以及鈣、鎂離子和緩沖系統。
鏈置換擴增其原理是基于在靶DNA兩端帶有被化學修飾的限制性核酸內切酶識別序列,核酸內切酶在其識別位點將鏈DNA打開缺口,DNA聚合酶延伸缺口3’端并替換下一條DNA鏈。
被替換下來的DNA單鏈可與引物結合并被DNA聚合酶延伸成雙鏈。該過程不斷反復進行,使靶序列被高效擴增。
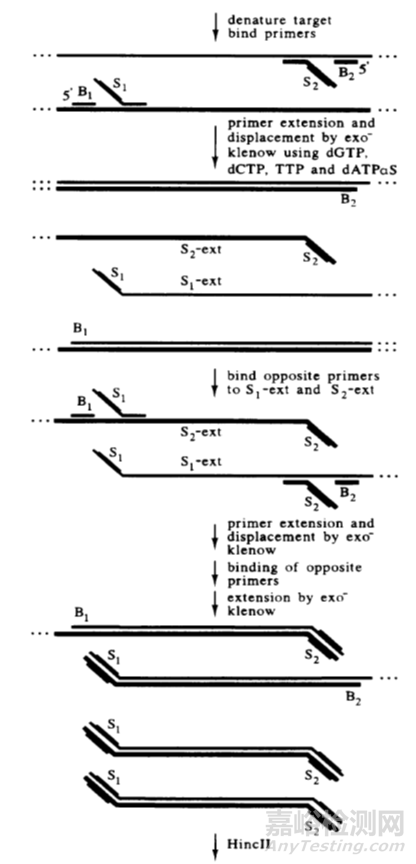
鏈置換擴增技術的優勢與不足
SDA 的優點:
擴增效率高,反應時間短,特異性強,不需要特殊的設備。
SDA 的不足:
產物不均一,在SDA循環中總要產生一些單、雙鏈產物,用電泳法檢測時必然會出現拖尾現象。
滾環核酸擴增
滾環核酸擴增(rolling circle amplification, RCA)是通過借鑒病原生物體滾環復制DNA的方式而提出的,指在恒溫下以單鏈環狀DNA為模板,在特殊的DNA聚合酶(比如Phi29)的作用下,進行滾環式DNA合成,實現目標基因的擴增。
RCA可分為線性擴增與指數擴增兩種形式,線性RCA的效率可達到105倍,而指數RCA的效率可達到109倍。
簡單區分,如下圖,線性擴增a只用1條引物,指數擴增b則有2條引物。

線性RCA 又稱為單引物RCA,一條引物結合到環狀DNA上,在DNA 聚合酶作用下被延伸,產物為單環長度數千倍的大量重復序列的線狀單鏈。
由于線性RCA的產物始終連接在起始引物上,所以信號易于固定是它的一大優勢。
指數RCA,也被稱作超分支擴增HRCA(Hyper branched RCA),在指數RCA中,一條引物擴增出RCA產物,第二條引物與RCA產物雜化并延伸,置換已經結合在RCA產物上的下游引物延伸鏈,反復進行延伸和置換,產生樹狀的RCA擴增產物。

以4BaseBio公司的TruePrime滾環擴增試劑盒為例,使用TthPrimPol引物酶和Phi29DNA聚合酶,實現了無需添加引物即可進行擴增。

滾環核酸擴增的優勢與不足
RCA 的優勢:靈敏度高,特異性好,易操作。
RCA 的不足:信號檢測時的背景問題。在RCA反應過程中未成環的鎖式探針和未結合探針的模板DNA或者RNA 可能產生一些背景信號。
依賴核酸序列的擴增技術
依賴核酸序列的擴增技術(nucleicacid sequence-based amplification, NASBA)是在PCR基礎上發展起來的一種新技術,是由1對帶有T7啟動子序列的引物引導的連續、等溫的核酸擴增技術,可以在2h左右將模板RNA擴增約109倍,比常規PCR法高1000倍,不需特殊的儀器。
該技術一出現就被用于疾病的快速診斷,目前有不少公司的RNA檢測試劑盒都用此方法。
盡管RNA的擴增也可以使用反轉錄PCR技術,NASBA相比則有自己的優勢:可以在相對恒溫的條件下進行,相對傳統的PCR技術更為穩定、準確。
反應在41攝氏度下,需要AMV(avian myeloblastosis virus)逆轉錄酶、RNA酶H、T7 RNA聚合酶和一對引物來完成。
其過程主要包括:
正向引物包含T7啟動子互補序列,反應過程中正向引物與RNA鏈結合,由AMV酶催化形成DNA-RNA雙鏈。
RNA酶H消化雜交雙鏈中的RNA,保留DNA單鏈。
在反向引物與AMV酶的作用下形成含有T7啟動子序列的DNA雙鏈。
在T7 RNA聚合酶的作用下完成轉錄過程,產生大量目的RNA。

NASBA的優勢:
(1)它的引物上帶有T7啟動子序列,而外來雙鏈DNA無T7啟動子序列,不可能被擴增,因此該技術具有較高的特異性和靈敏度。
(2)NASBA將反轉錄過程直接合并到擴增反應中,縮短了反應時間。
NASBA的劣勢:
(1)反應成分比較復雜。
(2)需要3種酶使得反應成本較高。
參考文獻:
1.姜蘇 , 李一榮,等溫擴增技術的原理及應用
2.彭濤,核酸等溫擴增技術及其應用
3.趙璐瑤等,基于環介導核酸等溫擴增的快檢技術研究
4.England biolabs, Loop Mediated Isothermal Amplification.
5.G.TerranceWalker etc. Strand displacement amplification isothermal, in vitro DNA amplification technique.
6.吳曉亮等,DNA的滾環擴增技術研究進展。
7.各公司官網。
03、下篇:測序技術
本篇作為分子診斷技術全解析的最后一篇,介紹一下測序技術。
基因測序技術始于1977年,sanger發明的DNA雙脫氧末端終止測序法拉開了序幕。Sanger在1958年和1980年因為胰島素測序和DNA測序而兩度獲得諾貝爾化學獎,是第四位兩度獲得諾貝爾獎以及唯一獲得兩次諾貝爾化學獎的人。
Sanger測序
Sanger測序采用四個雙脫氧核糖核酸(ddNTP)加入到正在合成的鏈上,由于雙脫氧核糖核酸少了一個氧原子,一旦被加入到DNA鏈上,反應即終止。
通過構建四個反應體系,加入AGCT四種雙脫氧核糖核酸,同時調節脫氧核糖核酸(dNTP)和ddNTP的相對濃度,可以使反應擴增得到幾百至上千堿基的終止產物。
隨后通過凝膠電泳對產物進行分離,分為四個泳道,每個泳道對應一種堿基,然后讀取條帶顯影結果。

該方法因準確率高,被稱為基因檢測的金標準,但是耗時長,成本高。
2001年人類基因組計劃就是采用sanger測序完成的,從1990年人類基因組計劃建立開始,全球的科學家歷時11年耗資30億美元完成。
進入21世紀后,隨著物理及化學技術的發展,開始采用相同激發波長但不同發射波長的熒光集團來標記ddNTP,ATGC對應不同的熒光基團產生不同顏色的光被計算機讀取,測序的速度和效率大大提高。
二代測序
第二代測序技術也稱高通量測序(high-throughput sequencing,HTS)技術,相對于一代測序,它可以實現大規模平行測序,基本原理是將基因組分割成短片段,對短片段測序再進行拼接。
對比第一代測序技術擁有著高通量、低成本等優勢,目前相同數據量的檢測,其成本約為一代測序技術的0.01%,極大地推動了測序技術在臨床檢測方面的應用。
2005年454公司基于焦磷酸測序法推出了Genome Sequencer 20 System(GS 20)系統,開啟了高通量測序的進程。2007年,羅氏公司收購了454,并推出了一系列性能更優的NGS系統,極大的提升測序通量和準確性。
盡管具有讀長優勢,但是測序通量和成本始終限制了454平臺的推廣,同樣數據量下成本約是illumina的100倍,因此羅氏在2016年底終止了454NGS測序相關的業務。
2006年Solexa公司推出了Genome Analyzer系統,包括DNA簇、橋式PCR和可逆阻斷等技術,這使得GA系統在高通量、低成本、應用范圍廣等方面具有明顯優點。2007年,Illumina公司收購了Solexa并發布二代測序儀。
二代測序經過這些年的發展已經步入成熟期,目前市場上根據測序技術可以可以把二代測序平臺分為4類:邊合成邊測序法(Illumina)、半導體測序法(ThermoFisher)、聯合探針錨定聚合測序法(華大智造)和焦磷酸測序法。
Illumina邊合成邊測序
Illumina測序的流程主要包括樣品制備,簇生成,測序,數據分析。
首先是樣本制備和建庫。用DNA或RNA抽提試劑盒提取核酸,然后用超聲波將其隨即打斷成90-250bp左右的長度或者控制全部DNA在一定長度范圍內。
為了后續的擴增和測序,需要在這些DNA片段加入特定的序列。如下圖,分別是與流動池引物互補結合的區域(P5、P7)、與Read 1和read 2測序引物結合的區域(Rd1SP,Rd2 SP)以及標簽序列區域Index。

添加完接頭序列的DNA合集稱為DNA文庫,這樣就完成了建庫,該步驟可以采用商業化的文庫制備試劑盒完成。
第二步是成簇Cluster Generation。
成簇是上述DNA片段被擴增的過程,該過程在流動池(Flow cell) 中完成。流動池是一種含有8個通道的厚玻璃片,每條通道中都隨即植入了能與文庫接頭P5或P7互補結合的短DNA片段。

首先引物和流動池的固定DNA片段互補配對,固定在通道表面,然后在DNA聚合酶作用下DNA鏈進行互補延伸形成DNA雙鏈。通過變性,其中的單鏈被洗脫,剩下的一條單鏈會與旁邊的固定接頭鏈接,形成單鏈橋。

同樣的,單鏈橋在DNA聚合酶作用下延伸配對形成雙鏈橋,通過變性形成2條單鏈,這兩條單鏈又分別與旁邊的固定引物結合,形成2個單鏈橋。重復這個循環,最終形成數百萬的DNA簇。

上述過程所有的DNA片段都會被擴增,擴增結束后,反向連會被切斷洗脫,只留下正向鏈,為防止互補結合重新形成單鏈橋,3‘端被封鎖。

第三步,測序。
首先,在流動池中加入熒光標記的dNTP和酶,由引物起始開始合成子鏈。由于dNTP存在 3’端疊氮基會阻礙子鏈延伸,因此每個循環只能測得一個堿基。合成完一個堿基后,洗掉多余的dNTP和酶,使用激光掃描獲得熒光信號。
隨后加入試劑將疊氮基團與熒光基團切除,然后流動池再通入熒光標記的dNTP和酶,由引物起始開始合成一個堿基。不斷重復這個過程,完成第一次讀取。
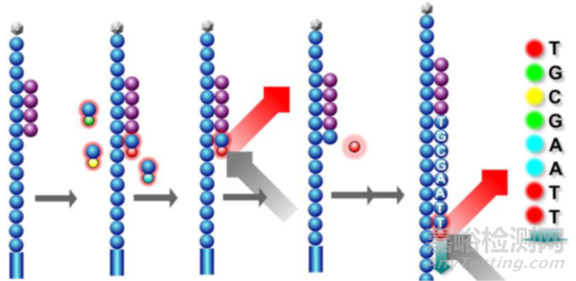
所有的DNA片段的一個堿基會被同時讀取,在大規模并行的過程中,機器讀取的圖像類似下面這樣:

同時,加入了不同的index來區分每個樣本及正負鏈。在完成第一次讀取后,復制出的鏈會被洗去,index片段引物被引入并與模板雜交,完成序列讀取后被洗去。這樣讀取到的序列與開始時已知的index比對后就可以給測得的序列貼上標簽,方便后續分析。
Paired-end測序已經是現在的主流,要完成雙末端測序,首先要將模板鏈3’去保護,模板折疊,index片段引入,在聚合酶參與下形成雙鏈橋,然后變性,恢復為單鏈,然后將正向鏈切除并洗去,留下反向鏈,與正向鏈類似,經過多個循環后完成讀取。
第四步,數據分析
測序完成后會產生數百萬個reads,基于在樣品準備時構建的index 分類來自不同樣本的序列。對于每個樣品來說,具有相似延伸的堿基被聚在一起。正向和反向read配對生成連續序列。這些序列通過與參考基因組匹配后,實現完整序列的構建。
Thermo Fisher半導體測序法
賽默飛的Ion Torrent平臺是基于半導體技術的高通量測序儀。該平臺使用了一種布滿小孔的高密度半導體芯片,一個小孔就是一個測序反應池,孔底部帶有感應器。

其測序的核心技術是利用半導體技術進行信息讀取,測序過程中每當有堿基結合,便會釋放H+,而氫離子會引起電勢的改變從而被檢測到。通過對氫離子的檢測并轉化為電信號,最終實現實時堿基判讀。
其測序的流程主要包括建庫,油包水PCR,測序,數據分析。
首先是建庫
與illumina 的 3’端帶突出 T 堿基粘性末端的接頭有所不同,建庫過程中DNA兩端加入的是平端接頭(P1)和X或A接頭,X接頭帶有index,A接頭不帶。

X 接頭帶Barcode 序列(近末端藍色序列),而A 接頭不帶Barcode 序列。
X 接頭的好處是可以把一個芯片的測序通量分配給多個文庫,測完序之后用Barcode 區分。
A 接頭的好處是直接測到樣本序列,這樣對于充分利用測序的讀長無疑是更好的。但是它的缺點是沒有Barcode,所以一張芯片只能放一個樣本。
AmpliSeq 是Ion Torrent 平臺上的建庫方案,它的核心是通過多重PCR 的方法,一次從樣本中把要測序的多個DNA 片段給擴增出來,然后轉化成文庫進行測序。

PCR 產物兩端20-30bp 堿基都是 PCR引物的序列,如果將其進行測序,則會浪費掉相當一部分測序讀長及數據量。因此在這個引物上特別設計了一種化學修飾,這種化學修飾可以被Fupa試劑所消化,而后的測序就可以盡可能多地測到樣本序列。
乳液PCR或油包水PCR
Ion Torrent 測序前需要把文庫結合到測序微珠上去,并且進行擴增,此種方法稱為油包水PCR,也稱Emulsion PCR(乳液PCR)。
EP 管中包含油相和水相,其中水相是核心,油相起到分隔作用。水相中包括文庫、引物、酶、MasterMix、測序微珠等PCR反應的主要成份。

測序微珠的直徑約1~2.4微米,每個油包水PCR都含有許多微珠,這些微珠的表面共價連接了許多PCR引物,與P1序列互補。
同時油包水PCR中的游離PCR引物,其序列與A/X接頭一致,且5‘端都標記了生物素。
把引物、酶、測序微珠等先在水相中混合,再加入油,混合形成乳濁液,油把水相分隔成一個個的小水滴。PCR反應后,微珠表面就會長出水滴內所含DNA 文庫的擴增拷貝。

然后加入帶有鏈霉親和素標記的磁珠與微珠進行混合。發生了PCR 的微珠由于其引物上帶有生物素,便會與磁珠結合;沒有發生PCR 的微珠由于沒有生物素,不會與磁珠結合。接著,用磁鐵吸附富集有效微珠,再清洗掉上清液中沒有PCR 的微珠,最后用洗脫液把微珠與磁珠分離開來,進行測序。
測序
測序主要發生在一張半導體芯片上,上面做了數以百萬、千萬計的小孔,每個小孔的既是測序微珠的容器,又同時是一個微型的PH 計。
每個小孔正好可以容納一個測序微珠,當DNA聚合酶把核苷酸聚合到延伸的DNA鏈上時,會釋放出一個氫離子,反應池中的PH發生改變,位于池下的離子感受器就會感受到信號,把化學信號直接轉化為數字信號,從而讀出DNA序列。

數據分析
把分別含A、C、G、T 四種 dNTP 的溶液,分別依次地流過芯片的表面。
舉例來說,流入的是dCTP 溶液,而模板上正好有一個G 堿基,就發生聚合反應,并產生電壓變化,而且會被記錄下來。如果流入的溶液與模板上的堿基不匹配,就不會發生聚合反應,也就沒有電壓變化,也就不會有堿基被記錄下來。
如果正好有 2 個一樣的堿基相鄰,一次就會有2 個堿基被聚合到DNA 鏈上,電壓變化值就會加倍,序列中2 個新的堿基被記錄下來。
華大智造聯合探針錨定聚合測序法
2013年3月18日,華大基因對CG(Complete Genomics)公司全額收購,開啟了華大測序儀之路。歷經多年的研發和改進,華大基因相繼推出的BGISEQ-500、BGISEQ-50、MGISEQ-200、MGISEQ-2000M、MGISEQ-T7等測序體系。
其測序平臺采用的是DNB(DNA Nanoball ,DNA納米球)測序技術,每個DNA的直徑約220-240nm。
主要步驟包括文庫制備,cPAS測序,數據分析等。
樣本準備和建庫
MGI文庫構建采用泡狀接頭及與之對應的擴增產物,有長接頭及短接頭,單端和雙端index。
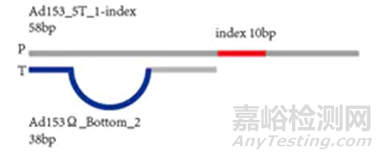
將文庫進行單鏈分離及環化處理后,以單鏈環狀DNA為模板,在DNA聚合酶作用下使用滾環擴增將單鏈環狀DNA擴增2-3個數量級,此時擴增產物被稱為DNB。
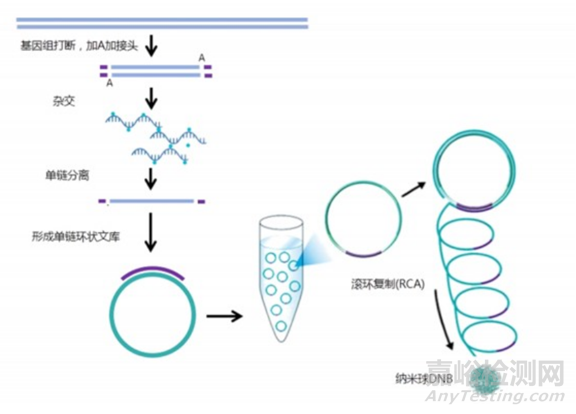
DNB經過DNB裝載技術固定在陣列化的硅芯片上形成納米芯片,由于一個DNB結合到芯片上的小孔后會排斥其他DNB結合,因此每個小孔僅容納一個DNB,保證了信號點間不會相互干擾。
測序
采用半導體加工工藝,在經過修飾的硅片表面形成結合位點陣列(直徑約200nm),實現DNA納米球的規則排列吸附,陣列位點的間距約700nm,每個位點只固定一個DNB,保證不同納米球之間的光信號不會互相干擾。
帶有熒光探針的Read1引物在DNB上匹配互補,隨后系統對光信號采集,得到待測序列。
MDA(multipledisplacement amplification)二鏈測序,隨機引物在多個位點與模板DNA結合,在Phi29DNA聚合酶作用下起始復制,沿著DNA模板合成DNA,同時取代模板互補鏈;被置換的互補鏈又變成新的模板進行后續擴增。完成第一鏈測序后,在該酶作用下形成第二鏈,通過DNA分子錨,進行第二鏈cPAS測序。

第三代測序技術
1.PacBio SMRT單分子測序技術
Pacific Biosciences于2004年成立,2010年納斯達克上市,2011年PacBioRS發布。
以PacBio測序為代表的第三代基因測序技術逐漸應用到多個科研領域。該平臺利于單分子實時測序技術,又稱作SMRT(Single Molecule Real-Time)測序,基于納米小孔的單分子讀取技術,無需擴增即可快速完成序列讀取。
建庫
因為測序讀長很長,因此可以制備大片段(3-10kb)文庫;不同于其他二代測序文庫,該文庫兩端分別連接環狀單鏈,單鏈兩端又與雙鏈正負鏈連接,得到類似啞鈴的結構,稱為SMRT Bell。
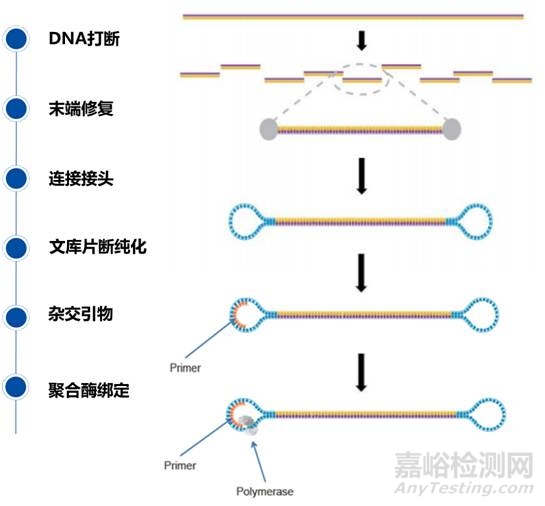
測序
SMRT Cell芯片含有數百萬個納米級的零模波導孔(zero-mode wave guides, ZMWs),ZMW是一個直徑只有10~50nm的孔,引物在模板的單鏈環部位退火后,這個雙鏈部位就可以結合到已固定在ZWM底部的聚合酶上。
每個ZMW都能夠包含一個DNA聚合酶及一條DNA樣品鏈進行單分子測序,4種dNTP帶有不同的熒光標記。當激光打在ZMW底部時,只能照亮很小的區域,DNA聚合酶就被固定在這個區域。

只有在這個區域內,堿基攜帶的熒光基團被激活從而被檢測到。當DNA合成進行時,下一個堿基進行延伸,上一個dNTP上的熒光基團就會被切除,保證了檢測的連續性。
不同的堿基會發出不同的光,此時根據光的波長及峰值便可判斷堿基類型。
2.納米孔測序技術
2005年,牛津納米孔科技有限公司(Oxford Nanopore Technologies)成立,致力于納米孔測序技術的商業轉化。2014年Nano space單分子測序技術發布。
牛津納米孔公司開發的納米單分子測序技術與以往的測序技術皆不同,它是基于電信號而不是光信號。
納米孔測序技術的核心是一種整合了多個跨膜通道蛋白(即納米孔蛋白)的多聚物膜。通過在膜兩側施加電壓從而產生穩定的穿過納米孔的電流。當有其他物體穿過納米孔時,會影響電流的大小,從而產生可識別的電信號的變化。
測序時,DNA雙鏈在馬達蛋白的牽引下解螺旋為單鏈DNA,并穿過納米孔蛋白(也叫Reader蛋白)。由于ATCG四種堿基結構和大小的差異,會使電流產生特征性離子電流變化,通過識別這種電信號的變化,從而達到讀取堿基序列的目的。

不同型號的納米孔測序儀原理非常一致,其核心均為納米孔測序芯片。常規的納米孔測序芯片整合512個測序通道,每個測序通道包含四個納米孔,官方數據顯示單張芯片不間斷測序48h,可產生20-30G的數據量。

二代測序的出現極大地解決了通量問題,在大幅提高測序速度和準確性的同時大大降低了測序成本,但閱讀長度相對較短。而以單分子測序為主要特征的三代測序,正朝著單分子、長讀長、低成本、小型化的方向發展,實現了測序領域的又一次變革。
參考文獻:
1.李金明,《高通量測序技術》
2.各公司官網

來源:Internet


